
Pourquoi Développer l’Analyse Cap de l’Expression génique?
Les recherches menées par Piero Carninci et Yoshihide Hayashizaki à la fin des années 1990, qui ont commencé avec la méthode cap trapper, l’utilisation du tréhalose, la méthode de normalisation/soustraction et un nouveau vecteur de clonage, ont ouvert la voie au développement de l’analyse Cap de l’expression des gènes. Avec cap trapper, des hybrides ADNc / ARNm sur toute la longueur sont isolés, et l’ARNm est chimiquement biotinylé sur la structure du capuchon et des billes magnétiques recouvertes de streptavidine capturent les hybrides. Cette avancée dans la série des principales technologies de l’ADN est décrite par Nature comme le jalon 5 (www.nature.com/milestones/miledna/full/miledna05.html Leur but dans le développement de CAGE était de créer une technologie permettant de cartographier de manière exhaustive la grande majorité des sites de départ de la transcription humaine et leurs promoteurs. En fait, la technologie permettant de profiler l’activité de la transcription des gènes à chaque site promoteur n’existait pas avant l’arrivée de CAGE. L’ARN messager (ARNm) représente un lien critique entre les informations codées dans les gènes individuels d’un génome et la composition protéique qui détermine le destin d’un organisme. Nous nous sommes demandé : quelles sont les régions génomiques, ou promoteurs, qui stimulent l’expression spécifique des gènes et de leurs ARN uniques ? En effet, des collections complètes d’ADNc ont montré que la plupart des gènes ont plus d’un site de départ de transcription et qu’il est donc assez difficile d’identifier les régions contrôlantes, c’est-à-dire les promoteurs, responsables de l’expression des différentes formes de transcriptions.
L’analyse de la complexité de ce processus de transfert d’informations, appelé « transcription », nécessite le développement d’outils moléculaires sophistiqués capables de saisir à la fois les aspects qualitatifs et quantitatifs de l’expression des gènes. Ainsi, avec CAGE, notre technologie originale de détection de démarrage de la transcription à l’échelle du génome, nous pouvons effectuer un profilage de l’expression génique à grande échelle avec une identification simultanée des sites de démarrage de la transcription (TSS) spécifiques au tissu, à la cellule et à la condition, y compris l’analyse de l’utilisation du promoteur. CAGE est basée sur la préparation et le séquençage de concatamères de balises d’ADN dérivant des 20 nucléotides initiaux des ARNM d’extrémité 5′, qui reflètent la concentration initiale d’ARNM dans l’échantillon analysé (fréquence des ARN).
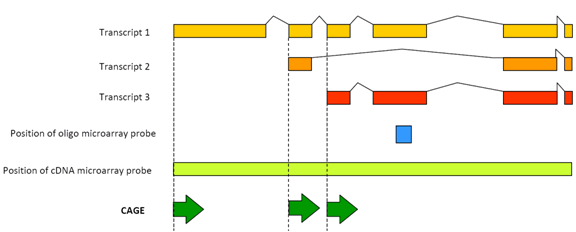
Figure 1: CAGE détecte l’activité transcriptionnelle de chaque transcription promotrice.
Les étiquettes de séquençage exprimées (EST) ont été utilisées dans les premiers stades du développement technologique pour identifier les éléments promoteurs en les alignant sur le génome humain. Cependant, ce processus est très coûteux en raison des coûts de traitement des ADNC physiques et du séquençage de Sanger. Une façon de surmonter ces problèmes est d’utiliser des technologies de marquage, qui ont été développées pour détecter des transcrits avec une sensibilité supérieure d’au moins un ordre de grandeur au séquençage EST, puis identifier de manière exhaustive les transcrits, identifier leurs promoteurs et les corréler avec des profils d’expression en comptant les étiquettes comme mesure numérique de l’expression génique.
Ces séquences (également appelées » tags ») sont ensuite alignées sur les séquences du génome par des procédures de calcul simples (appelées BLAST) et comptées, ce qui donne une mesure de la fréquence d’expression de l’ARN. Comme ces étiquettes de séquence identifient les sites de départ de la transcription de l’ARN, elles identifient également les séquences du génome proches de ces sites de départ. Les régions voisines sont les régions promotrices principales, qui sont des séquences génomiques qui provoquent la transcription des gènes dans les nombreuses conditions différentes rencontrées dans de nombreux organismes complexes, de la souris à l’homme.
Attributs de CAGE
CAGE présente de grands avantages par rapport aux techniques classiques de détection d’expression à base de microréseaux. En effet, en identifiant le promoteur à l’origine de la transcription de l’ARN dans chaque phénomène biologique, tissu, cellule, etc. nous pouvons identifier les éléments régulateurs de l’ADN spécifiques à chaque phénomène biologique en regardant les séquences qui sont dans les promoteurs des isoformes d’ARN exprimées dans les échantillons analysés. Les promoteurs contiennent des séquences spécifiques, ou sites de liaison des facteurs de transcription (TFB), qui sont reconnus par leurs protéines de liaison appelées facteurs de transcription (TF) et favorisent, ou encore répriment, la transcription. À l’aide de méthodes de calcul, nos chercheurs analysent des promoteurs ayant des profils d’expression similaires pour leurs TFB, puis identifient les TF responsables de la sortie transcriptionnelle du génome. En comptant le nombre d’étiquettes de CAGE pour chaque promoteur au sein d’un gène, nous pouvons maintenant déterminer non seulement le niveau d’expression de l’ARN (il s’agit d’une détection numérique de la fréquence) mais, surtout, aussi à partir de laquelle des différents promoteurs alternatifs l’ARN est transcrit.
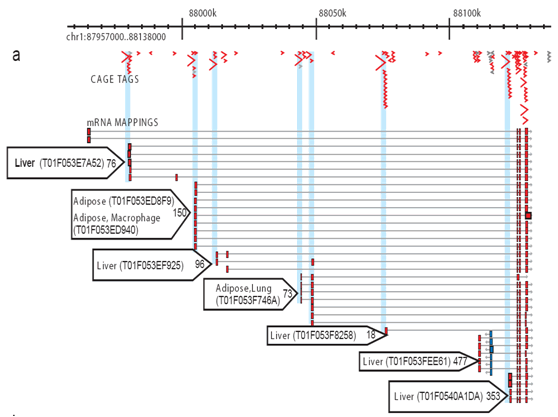
Figure 2: CAGE permet le profilage complet des activations à chaque site promoteur. Pour chaque bibliothèque, un certain nombre d’étiquettes en CAGE sont séquencées et alignées sur le génome afin de mesurer l’activité transcriptionnelle spécifique de chaque promoteur et de distinguer la contribution de chaque promoteur. Cet exemple simplifié montre les promoteurs du noyau adipeux et hépatique. Minuscules flèches bleues: étiquettes de CAGE individuelles; flèches rouges: préférence d’utilisation du promoteur pour les tissus; boîtes rouges: régions du promoteur principal.
Comme mentionné, CAGE utilise le cap-trapping comme première étape pour capturer les extrémités 5′ des ADNc, qui sont ensuite transformées en séquence courte (tags) de 20 à 27 nt correspondant à l’ARNm TSSs,. Nous avons produit des millions de balises de CAGE de souris et d’humains en utilisant des balises de CAGE concaténées avec séquençage Sanger, jusqu’à ce que nous passions récemment à deepCAGE, pour lequel nous utilisons le séquençage de deuxième génération.
Jusqu’en 2006, nous utilisions des bibliothèques de CAGES sur notre pipeline de séquençage RISA d’origine, qui a été construit à la fin des années 90 et comprenait le seul séquenceur capillaire avec un réseau de 384 capillaires que nous avons développé en collaboration avec la société Shimadzu.
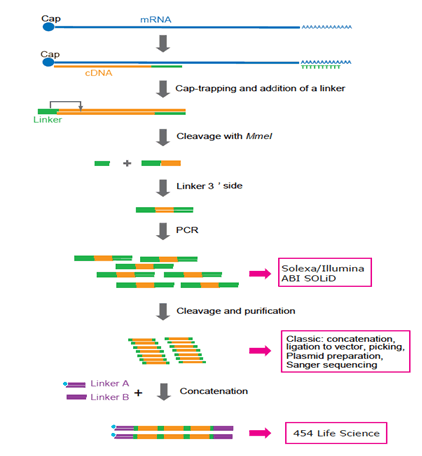
Figure 3 : Représentation du protocole de préparation de CAGE adapté aux différentes plateformes. Maintenant, Solexa et Illumina sont préférés. 454 Sciences de la vie (système FLX) n’est plus utilisé car la concaténation nécessite des cycles PCR supplémentaires et une manipulation compliquée. À l’avenir, la technologie de séquençage à molécule unique sera préférée car la PCR pourrait ne pas être nécessaire.
Bien que la technologie de marquage et de séquençage ait été développée à l’aide de la méthode SAGE (serial analysis of gene analysis), CAGE est unique car elle repose sur le principe du profilage de l’extrémité 5′ des ARN portant un site de cap – tous les ARNM et une grande fraction des ARN non codants. Nous avons développé des étiquettes à CAGE longue de 27 nt pour augmenter l’efficacité de la cartographie.