
Warum eine Cap-Analyse der Genexpression entwickeln?
Die Forschung von Piero Carninci und Yoshihide Hayashizaki in den späten 1990er Jahren, die mit der Cap-Trapper-Methode, der Verwendung von Trehalose, der Normalisierungs- / Subtraktionsmethode und einem neuen Klonierungsvektor begann, legte den Grundstein für die Entwicklung der Cap-Analyse der Genexpression. Mit Cap Trapper werden cDNA / mRNA-Hybride in voller Länge isoliert, und die mRNA wird auf der Cap-Struktur chemisch biotinyliert und Streptavidin-beschichtete Magnetperlen fangen die Hybride ein. Dieser Fortschritt in der Reihe der wichtigsten DNA-Technologien wird von Nature als Meilenstein 5 beschrieben.www.nature.com/milestones/miledna/full/miledna05.html ). Ihr Ziel bei der Entwicklung von CAGE war es, eine Technologie zur umfassenden Kartierung der überwiegenden Mehrheit der menschlichen Transkriptionsstartstellen und ihrer Promotoren zu schaffen. Tatsächlich existierte die Technologie zum Profilieren der Aktivität der Gentranskription an jeder Promotorstelle erst, als CAGE auf die Bühne kam. Messenger-RNA (mRNA) stellt eine kritische Verbindung zwischen den Informationen dar, die in einzelnen Genen eines Genoms kodiert sind, und der Proteinzusammensetzung, die das Schicksal eines Organismus bestimmt. Wir haben uns gefragt: Was sind die genomischen Regionen oder Promotoren, die die spezifische Expression von Genen und ihren einzigartig unterschiedlichen RNAs steuern? In der Tat haben cDNA-Sammlungen in voller Länge gezeigt, dass die meisten Gene mehr als eine Transkriptionsstartstelle haben, und daher ist es ziemlich schwierig, die kontrollierenden Regionen, dh Promotoren, zu identifizieren, die für die Expression der verschiedenen Formen von Transkripten verantwortlich sind. Die Analyse der Komplexität dieses Informationstransferprozesses, der als ‚Transkription‘ bezeichnet wird, erfordert die Entwicklung hochentwickelter molekularer Werkzeuge, die sowohl die qualitativen als auch die quantitativen Aspekte der Genexpression erfassen können. So können wir mit CAGE, unserer ursprünglichen genomweiten Transkriptionsstartdetektionstechnologie, ein hochpräzises Genexpressionsprofil mit gleichzeitiger Identifizierung der gewebe- / Zell- / zustandsspezifischen Transkriptionsstartstellen (TSS) einschließlich der Promotornutzungsanalyse durchführen. CAGE basiert auf der Herstellung und Sequenzierung von Concatameren von DNA-Tags, die aus den anfänglichen 20 Nukleotiden von 5′-End-mRNAs stammen und die ursprüngliche Konzentration von mRNA in der analysierten Probe widerspiegeln (RNA-Frequenz).
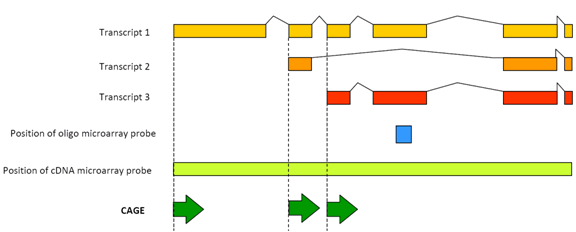
Abbildung 1: CAGE erkennt die Transkriptionsaktivität jedes Promotortranskripts.Exprimierte Sequenzierungs-Tags (ESTs) wurden in frühen Stadien der Technologieentwicklung verwendet, um Promotorelemente zu identifizieren, indem sie auf das menschliche Genom ausgerichtet wurden. Dieses Verfahren ist jedoch aufgrund der Kosten für die Handhabung physikalischer cDNAs und die Sanger-Sequenzierung sehr teuer. Eine Möglichkeit, diese Probleme zu überwinden, besteht darin, Tagging-Technologien zu verwenden, die entwickelt wurden, um Transkripte mit einer Empfindlichkeit zu detektieren, die mindestens eine Größenordnung größer ist als die EST-Sequenzierung, dann Transkripte erschöpfend zu identifizieren, ihre Promotoren zu identifizieren und sie mit Expressionsprofilen zu korrelieren, indem die Tags als digitales Maß für die Genexpression gezählt werden.Diese Sequenzen (auch „Tags“ genannt) werden dann durch einfache Rechenverfahren (BLAST genannt) auf Genomsequenzen ausgerichtet und gezählt, was eine Messung der Häufigkeit der RNA-Expression ergibt. Da diese Sequenz-Tags die Startstellen der RNA-Transkription identifizieren, identifizieren sie auch die Genomsequenzen in der Nähe dieser Startstellen. Die benachbarten Regionen sind die Kernpromotorregionen, bei denen es sich um genomische Sequenzen handelt, die dazu führen, dass Gene unter den vielen verschiedenen Bedingungen transkribiert werden, die in vielen komplexen Organismen, von Mäusen bis zu Menschen, auftreten.
Attribute von CAGE
CAGE hat große Vorteile gegenüber klassischen Microarray-basierten Expressionsdetektionstechniken. In der Tat durch Identifizierung des Promotors, der die RNA-Transkription in jedem biologischen Phänomen, Gewebe, Zelle usw. verursacht. wir können die DNA-regulatorischen Elemente identifizieren, die für jedes biologische Phänomen spezifisch sind, indem wir die Sequenzen betrachten, die sich in den Promotoren der RNA-Isoformen befinden, die in den analysierten Proben exprimiert werden. Promotoren enthalten spezifische Sequenzen oder Transkriptionsfaktor-Bindungsstellen (TFBS), die von ihren Bindungsproteinen, den sogenannten Transkriptionsfaktoren (TF), erkannt werden und die Transkription fördern oder alternativ unterdrücken. Mit computergestützten Methoden analysieren unsere Forscher Promotoren mit ähnlichen Expressionsprofilen für ihre TFBS und identifizieren dann die TFs, die für die transkriptionelle Ausgabe des Genoms verantwortlich sind. Indem wir die Anzahl der CAGE-Tags für jeden Promotor innerhalb eines Gens zählen, können wir jetzt nicht nur das RNA-Expressionsniveau bestimmen (dies ist eine digitale Erkennung der Häufigkeit), sondern vor allem auch, von welchem der verschiedenen alternativen Promotoren die RNA transkribiert wird.
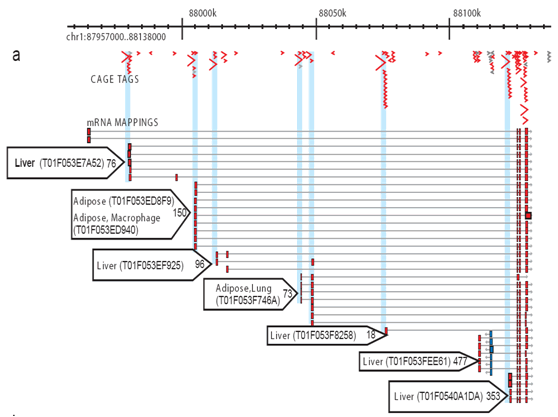
Abbildung 2: CAGE ermöglicht die umfassende Profilierung von Aktivierungen an jeder Promotorstelle. Für jede Bibliothek werden eine Reihe von CAGE-Tags sequenziert und an das Genom angepasst, so dass die spezifische Transkriptionsaktivität an jedem Promotor gemessen und der Beitrag jedes Promotors unterschieden werden kann. Dieses vereinfachte Beispiel zeigt Fett- und Leberkernpromotoren. Winzige blaue Pfeile: individuelle KÄFIG-Tags; rote Pfeile: Promoter-Verwendungspräferenz für Gewebe; rote Kästchen: Kernpromotorregionen.
Wie bereits erwähnt, verwendet CAGE Cap-Trapping als ersten Schritt, um die 5′-Enden der cDNAs zu erfassen, die dann in kurze Sequenzen (Tags) von 20 bis 27 nt umgewandelt werden, die den mRNA-TSSs entsprechen, . Wir haben Millionen von Käfigetiketten für Mäuse und Menschen mit verketteten Käfigetiketten mit Sanger-Sequenzierung hergestellt, bis wir kürzlich zu deepCAGE gewechselt sind, für das wir Sequenzierung der zweiten Generation verwenden.
Bis 2006 führten wir CAGE-Bibliotheken auf unserer ursprünglichen RISA-Sequenzierungspipeline aus, die Ende der 90er Jahre gebaut wurde und den einzigen Kapillarsequenzer mit einem Array von 384 Kapillaren enthielt, den wir in Zusammenarbeit mit der Shimadzu Corporation entwickelt hatten.
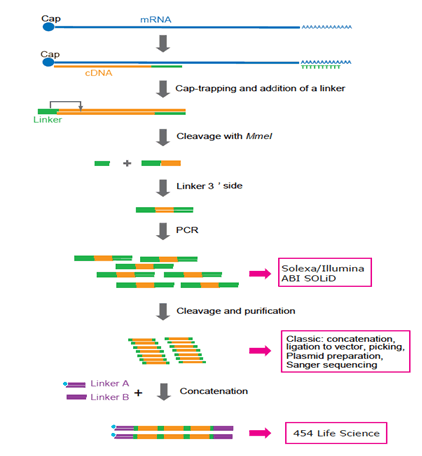
Abbildung 3: Darstellung des an verschiedene Plattformen angepassten Käfigvorbereitungsprotokolls. Jetzt werden Solexa und Illumina bevorzugt. 454 Life Sciences (FLX-System) wird nicht mehr verwendet, da die Verkettung zusätzliche PCR-Zyklen und komplizierte Manipulationen erfordert. In Zukunft wird die Einzelmolekülsequenzierungstechnologie bevorzugt, da die PCR möglicherweise nicht erforderlich ist.
Obwohl die Tagging- und Sequenzierungstechnologie unter Verwendung der SAGE-Methode (Serial Analysis of Gene Analysis) entwickelt wurde, ist CAGE einzigartig, da es auf dem Prinzip der Profilierung des 5′-Endes von RNAs basiert, die eine Cap–Site tragen – alle mRNAs und ein großer Teil der nicht codierenden RNAs. Wir haben entwickelt 27-nt lange KÄFIG tags zu erhöhen die mapping effizienz.