
varför utveckla Cap-analys av genuttryck?
forskning av Piero Carninci och Yoshihide Hayashizaki i slutet av 1990-talet, som började med cap trapper-metoden, användning av trehalos, normaliserings – /subtraktionsmetoden och en ny kloningsvektor, satte scenen för utveckling av Cap-analys av genuttryck. Med cap trapper isoleras cDNA/mRNA-hybrider i full längd och mRNA biotinyleras kemiskt på cap-strukturen och streptavidin-belagda magnetiska pärlor fångar hybriderna. Detta framsteg i serien av stora DNA-tekniker beskrivs av naturen som milstolpe 5 (www.nature.com/milestones/miledna/full/miledna05.html). deras syfte med att utveckla CAGE var att skapa en teknik för omfattande kartläggning av de allra flesta mänskliga transkriptionsstartplatser och deras promotorer. Faktum är att tekniken för att profilera aktiviteten för gentranskription vid varje promotorplats inte existerade förrän CAGE kom på scenen. Messenger RNA (mRNA) representerar en kritisk länk mellan informationen som kodas i enskilda gener på ett genom och proteinmakeupen som bestämmer en organisms öde. Vi frågade oss själva: Vilka är de genomiska regionerna, eller promotorerna, som driver det specifika uttrycket av gener och deras unikt olika rna? Faktum är att cDNA-samlingar i full längd har visat att de flesta gener har mer än en transkriptionsstartplats och det är därför ganska svårt att identifiera de kontrollerande regionerna, det vill säga promotorer, som är ansvariga för uttrycket av de olika formerna av transkript. att analysera komplexiteten i denna informationsöverföringsprocess, kallad ’transkription’, kräver utveckling av sofistikerade molekylära verktyg som kan fånga både de kvalitativa och kvantitativa aspekterna av genuttryck. Så med CAGE, vår ursprungliga genomövergripande transkriptionsstartdetekteringsteknik, kan vi göra hög genomgående genuttrycksprofilering med samtidig identifiering av vävnads – /cell – /tillståndsspecifika transkriptionsstartplatser (TSS), inklusive promotoranvändningsanalys. CAGE är baserat på beredning och sekvensering av konkatamrar av DNA-taggar som härrör från de initiala 20 nukleotiderna från 5′ end mRNA, vilket återspeglar den ursprungliga koncentrationen av mRNA i det analyserade provet (RNA-frekvens).
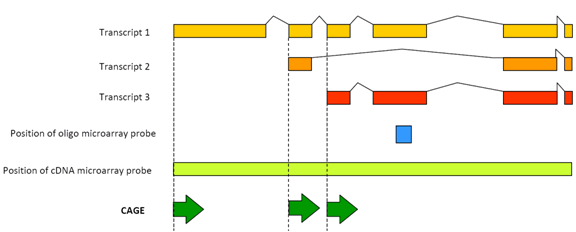
Figur 1: CAGE upptäcker transkriptionsaktiviteten för varje promotoravskrift.
uttryckta sekvenseringstaggar (est) användes i tidiga stadier av teknikutveckling för att identifiera promotorelement genom att anpassa dem till det mänskliga genomet. Denna process är dock mycket dyr på grund av kostnaderna för hantering av fysiska cDNA och Sanger-sekvensering. Ett sätt att övervinna dessa problem är att använda taggteknik, som har utvecklats för att upptäcka transkript med en känslighet som är minst en storleksordning större än EST-sekvensering, sedan uttömmande identifiera transkript, identifiera deras promotorer och korrelera dem med uttrycksprofiler genom att räkna taggarna som ett digitalt mått på genuttryck.dessa sekvenser (även kallade ”taggar”) anpassas sedan till genomsekvenser genom enkla beräkningsprocedurer (kallad BLAST) och räknas, vilket ger en mätning av frekvensen av RNA-uttryck. Eftersom dessa sekvenstaggar identifierar startplatserna för RNA-transkriptionen identifierar de också genomsekvenserna nära dessa startplatser. Grannregionerna är kärnpromotorregionerna, som är genomiska sekvenser som får gener att transkriberas vid de många olika förhållanden som påträffas i många komplexa organismer, från möss till människor.
attribut för bur
bur har stora fördelar jämfört med klassiska mikroarraybaserade uttrycksdetekteringstekniker. Faktum är att genom att identifiera promotorn som orsakar RNA-transkription i varje biologiskt fenomen, vävnad, cell etc. vi kan identifiera DNA-reglerande element som är specifika för varje biologiskt fenomen genom att titta på sekvenserna som finns i promotorerna för RNA-isoformerna som uttrycks i de analyserade proverna. Promotorer innehåller specifika sekvenser, eller transkriptionsfaktorbindningsställen (tfbs), som känns igen av deras bindande proteiner som kallas transkriptionsfaktorer (TF) och främjar eller alternativt undertrycker transkription. Med hjälp av beräkningsmetoder analyserar våra forskare promotorer som har liknande uttrycksprofiler för sina TFBS och identifierar sedan TFs som är ansvariga för transkriptionsutgången från genomet. Genom att räkna antalet BURTAGGAR för varje promotor inom en gen kan vi nu bestämma inte bara RNA-uttrycksnivån (detta är en digital detektering av frekvens) utan, viktigare, också från vilken av de olika alternativa promotorerna RNA transkriberas.
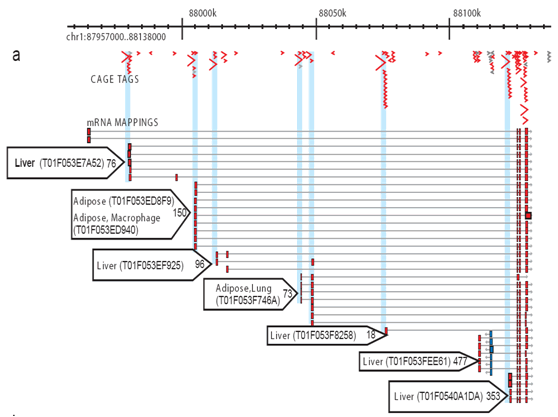
Figur 2: CAGE tillåter omfattande profilering av aktiverar vid varje promotor plats. För varje bibliotek sekvenseras ett antal BURTAGGAR och anpassas till genomet så att den specifika transkriptionsaktiviteten hos varje promotor kan mätas och varje promotors bidrag särskiljas. Detta förenklade exempel visar fett-och leverkärnpromotorer. Små blå pilar: enskilda bur taggar; röda pilar: promotor användning preferens för vävnader; röda rutor: core promotor regioner.
som nämnts, CAGE använder cap-fångst som det första steget för att fånga de 5′ ändarna av cDNAs, som sedan omvandlas till kort sekvens (taggar) av 20 till 27 nt motsvarar mRNA TSSs, . Vi har producerat miljontals mus-och mänskliga BURTAGGAR med sammanlänkade BURTAGGAR med Sanger-sekvensering, tills vi nyligen flyttade till deepCAGE, för vilken vi använder andra generationens sekvensering. fram till 2006 körde vi BURBIBLIOTEK på vår ursprungliga risa-sekvenseringsrörledning, som byggdes i slutet av 90-talet och inkluderade den enda kapillärsekvenseraren med en rad 384 kapillärer som vi utvecklade i samarbete med Shimadzu Corporation.
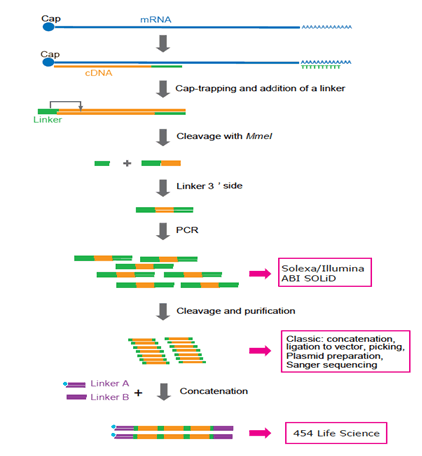
Figur 3: Representation av BURBEREDNINGSPROTOKOLL anpassat till olika plattformar. Nu föredras Solexa och Illumina. 454 Life Sciences (FLX-system) används inte längre eftersom sammanfogning kräver ytterligare PCR-cykler och komplicerad manipulation. I framtiden kommer enmolekylsekvenseringsteknik att föredras eftersom PCR kanske inte krävs.
även om märkning och sekvenseringsteknik har utvecklats med hjälp av serial analysis of gene analysis (SAGE)-metoden, är CAGE unik eftersom den bygger på principen att profilera 5′ – änden av RNA som bär en cap-site-all mRNA och en stor del av de icke-kodande RNA. Vi har utvecklat 27-nt långa BURTAGGAR för att öka kartläggningseffektiviteten.