
Introdução
A completar sequências do genoma de vários organismos, incluindo mamíferos, têm recentemente se tornou disponível como uma consequência de rápidos avanços na tecnologia de sequenciamento de DNA. No entanto, particularmente em mamíferos, a análise de transcrições ainda desempenha um papel fundamental na ponte entre o genoma e o proteoma. Isso ocorre principalmente porque, atualmente, não podemos prever com precisão as estruturas de transcrições derivadas de um determinado gene apenas da informação genômica. Assim, como um método para a análise de transcrições, a construção da biblioteca cDNA é crucial, mesmo na era pós-sequenciação do genoma. Embora a clonagem cDNA de genes de interesse pela PCR tenha fornecido uma via alternativa simplificada para analisar estruturas de transcrição sem a construção da biblioteca cDNA, a construção de uma biblioteca cDNA é a abordagem de escolha quando um grande número de cDNAs de uma única fonte de mRNA devem ser analisados. Até à data, tem sido feito um grande esforço para desenvolver um método para a preparação de bibliotecas cDNA de alta qualidade (1-4). Em contraste, a questão de como preparar uma biblioteca cDNA de alta qualidade a partir de um pequeno pool de RNAs não foi tão ativamente abordada. No entanto, este tornou-se um objectivo prioritário porque os investigadores estão frequentemente interessados em genes hipotéticos que se prevê serem expressos apenas em certos tipos de células ou tecidos em determinadas condições, tais como as observadas em amostras patológicas.
tem havido uma série de relatórios descrevendo o uso de pequenas quantidades de RNA fonte para gerar cDNA amplificado ou cRNA, a partir do qual alvos para análise de microarray podem ser preparados (5-15). Seu objetivo final é obter RNA de comprimento total, não-População tendenciosa, que é altamente representativo do ARNm original. Para realizar isso, estes métodos geralmente adotam a transcrição PCR ou in vitro pela polimerase de RNA T7 para amplificar o ARNm original na forma de cDNA ou cRNA, respectivamente. Embora a comparação de perfis de expressão seja uma das abordagens mais eficientes para investigar diferenças nos estados fisiológicos de células ou tecidos, a caracterização estrutural de transcritos (por exemplo, identificação de padrões alternativos de splicing, local de início de transcrição, e local de terminação de transcrição) não pode ser feita por tal análise de microarray. A análise sequencial de cada transcrição é inevitável nestes casos.
desenvolvemos um método que permite que uma pequena quantidade de RNA inicial seja usada para construir uma biblioteca cDNA adequada para clonagem de genes e análise de sequenciamento abrangente. O método marca as extremidades 3′ e 5′ das cDNAs de primeira rodada com sequências promotoras de Fages T7 e SP6, respectivamente, para minimizar os efeitos de viés de tamanho durante a amplificação. Após a amplificação do cRNA de duas voltas, convertemos o cRNAs amplificado em cDNAs, clonamos esses produtos em um plasmídeo, e então avaliamos a biblioteca cDNA resultante em comparação com a construída por um método convencional (4,16,17).
materiais e métodos
amplificação de cRNA
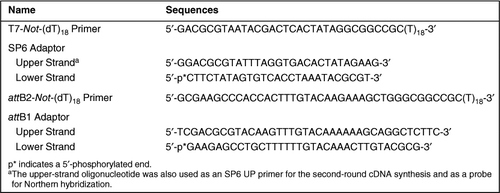
as sequências de oligonucleótidos utilizadas neste estudo são apresentadas na Tabela 1. Para estabelecer o protocolo, usamos 1 µg de RNA total Preparado a partir do cérebro do rato ICR (ratinho macho de 8 semanas de idade) como um modelo. Incubou-se uma mistura (10 µL) de 1 µg de RNA total e 100 pmol T7-não-(dT)18 primer a 70°C durante 10 minutos e depois arrefeceu no gelo. A síntese e ligação do adaptador cDNA de cadeia dupla foram realizadas seguindo os protocolos do sistema plasmídeo superscript® (Invitrogen, Carlsbad, CA, EUA) (4,16,17), com pequenas modificações. Resumidamente, para a síntese de cDNA da primeira cadeia, foram adicionados à mistura primária RNA/T7-não-(dT) 18 µl 10 mM dNTPs, 1 µL (40 U) RNaseOUT™ (Invitrogen), 1 µL de água desnaturada e incubada a 37°C durante 3 minutos a iniciadores de anneal. Em seguida, 2 µL (400 U) SUPERSCRIPT III RNase h-transcriptase reversa (Invitrogen) foram adicionados à mistura de reação, e a temperatura foi ajustada para 50°C para iniciar a síntese de cDNA da primeira cadeia. Após 1 h, segunda-strand cDNA a síntese foi realizada como descrito anteriormente por Gubler e Hoffman (18); para o primeiro-strand cDNA mistura, 91 µL de água, 30 µL de 5× segunda-strand buffer (Invitrogen), 3 µL de 10 mM de dNTPs, 1 µL (10 U) Escherichia coli DNA ligase, 4 µL (40 U) E. coli DNA polimerase, e 1 µL (2 U) E. coli RNase H foram adicionados, e a mistura foi incubada a 16°C por 2 h. cDNA termini, em seguida, foram de fim-polido com 10 U de T4 DNA polimerase (Invitrogen) a 16°C por 5 min, e a reação foi interrompida pela adição de 10 µL 0,5 M etilenodiamina tetraacetic acid (EDTA). Os cDNAs resultantes foram purificados por extração com fenol/clorofórmio / álcool isoamílico (25:24:1), seguido de precipitação de etanol como anteriormente descrito (17), exceto para a utilização de 1 µg de tRNA de levedura em vez de ácido poliadenílico como portador. O cDNA precipitado foi dissolvido em 50 µL TE (solução Tris-HCl de 10 mM, pH 8,0 e EDTA de 1 mM), misturado com 30 µL de uma solução de 20% (M/v) de polietilenoglicol (PEG) 6000/2.5 M de NaCl e, em seguida, incubado no gelo durante 1 h (17). Após a incubação, a mistura de cDNA foi centrifugada a 18.000× g A 4°C durante 15 minutos. O sedimento cDNA resultante foi enxaguado duas vezes com etanol a 70%, seco e ressuspenso em 30 µL de água. Os cDNAs purificados foram ligados com 500 Adaptadores SP6 pmol (Tabela 1) Usando 5 U T4 DNA ligase (Invitrogen) em um volume de reação de 50 µL a 16°C durante a noite. A mistura ligada ao adaptador foi diluída duas vezes com água, e então cDNAs foram purificados usando uma coluna DNAclear™ (Ambion, Austin, TX, EUA). O eluato (em 16 µL de água) foi usado como um modelo para a síntese de cRNA. Utilizando o Kit MEGAscript® T7 (Ambion), as cRNAs foram sintetizadas a 37°C durante a noite num volume de reacção de 40 µL. Após o modelo cDNAs ser degradado por 4 U DNase I, as cRNAs sintetizadas foram purificadas usando um Mini Kit RNeasy® (QIAGEN, Valencia, CA, EUA) e eluídas com 100 µL de água. A concentração do cRNA foi determinada por absorção ultravioleta (UV). Para a amplificação de cRNA de segunda fase, foram utilizados como template e primer 2 µg do cRNA sintetizado e 100 pmol SP6 UP primer (igual ao oligonucleótido de cadeia superior do adaptador SP6 mostrado na Tabela 1), respectivamente. A síntese de cDNA de segunda rodada foi realizada como na transcrição reversa original do RNA fonte, exceto que o recozimento do primer SP6 para cima foi realizado a 50 ° C durante 3 minutos. Após a purificação do cDNA de cadeia dupla obtido através de uma coluna DNAclear, 0,5 µg do cDNA foi usado como um modelo para a próxima amplificação CRNA assistida por polimerase SP6. Utilizando o Kit de MEGAscript SP6 (Ambion), a síntese de cRNA foi realizada a 37°C durante 6 H. Após o modelo cDNA ter sido degradado usando a DNase I, as cRNAs sintetizadas foram purificadas conforme descrito acima.
cDNA Library Construction
Dois microgramas da resultante cRNAs foram submetidos a duas vezes irrecuperáveis síntese de cDNA utilizando 100 pmol attB2-Não-(dT)18 de primers (Tabela 1). A síntese do cDNA, o end-blunting, a purificação e os passos do adaptador-ligação foram realizados como nas sínteses cDNA anteriores, exceto que o adaptador attB1 (Tabela 1, 500 pmol) foi ligado aos cDNAs de dupla cadeia. Após o attB1 adaptador-ligated cDNAs foram purificados por sucessivos tratamentos de fenol/clorofórmio/isoamyl extração do álcool, etanol precipitação, e PEG/NaCl a precipitação nesta ordem, foram dissolvidos em 50 µL de TE e tratado com RNase A (a uma concentração final de 10 µg/mL; Invitrogen) para degradar contaminados RNA a 37°C por 30 min. A mistura de reacção foi novamente purificada por extracção com fenol/clorofórmio/álcool isoamílico, seguida de precipitação de etanol e precipitação de PEG / NaCl. As cDNAs resultantes foram dissolvidas em 15 µL TE, e a sua quantidade foi estimada a partir da intensidade de coloração fluorescente após electroforese em gel de agarose (4). Os cDNAs ligados attB1 (36 ng) foram submetidos a uma reacção de recombinação in vitro (sistema Gateway®; Invitrogen) com 250 ng de vector dador attP-pSP73, tal como descrito anteriormente (4, 16, 17). Em resumo, o vetor dador atb1-ligado e attP-pSP73 foram misturados e incubados num volume de reação de 10 µL contendo 2 µL 5× BP Clonase™ tampão de reação e 2 µL BP Clonase (ambos de Invitrogen) a 25°C durante a noite. A mistura de cDNA foi tratada com 2 µg de proteinase K (Invitrogen) a 37°C durante 10 minutos para atenuar a reacção e extraída com fenol/clorofórmio/álcool isoamílico, seguida de precipitação de etanol com 2 µg de tRNA levedura. O cDNA precipitado foi dissolvido em 10 µL de água, e 1 µL da mistura foi utilizado para a transformação de células de E. coli ElectroMAX™ DH10B™ (Invitrogen). Após a titulação da biblioteca cDNA resultante, os plasmídeos cDNA foram recuperados de aproximadamente 1.000.000 colônias por um método de dodecilsulfato de sódio alcalino (SDS), como descrito anteriormente (4,19). Os clones cDNA resultantes estavam em uma forma de plasmídeos Carregando locais attL. Uma vez que os plasmídeos que transportam locais attB são necessários em algumas aplicações, estes plasmídeos foram convertidos em aqueles que transportam locais attB como descrito anteriormente (16). Esta biblioteca cDNA foi consequentemente designada como MB-AL (biblioteca derivada de cRNA amplificada pelo mouse brain).
da mesma forma como descrito para MB-AL, construímos uma biblioteca cDNA a partir de RNA não-amplificado (RNA total do cérebro de 100 µg do rato) e designámo-la como MB-CL (biblioteca construída convencionalmente pelo cérebro do rato).
Examination of Size-Bias Effect on Amplified cRNAs
To examine the size-bias effect on cRNAs, the first-round cRNAs transcribed by T7 RNA polymerase and the second-round cRNAs transcribed by SP6 RNA polymerase were run on 1,0% agarose gel (1 µg cada) and then blotted on nylon membrane (Biodyne® B Nylon Membrane; PALL, East Hills, NY, USA). Eles foram hibridizados com 32P-rotulado SP6 UP primer(Tabela 1) e não-(dT)18 (5′-GCGCGCTTTTTTTTTTTTTTTTTTTTTTTTTTTTT-3′) oligonucleótido, respectivamente. Dez picomoles de cada sonda foram rotulados com ATP (aproximadamente 3000 Ci/mmol).; Amersham Biosciences, Piscataway, NJ, USA) by using T4 polynucleotide kinase (Takara, Kyoto Japan). Após hibridação nocturna em tampão GMC (20) a 45°C, estas membranas foram lavadas extensivamente em solução salina de 1× citrato padrão (SSC)/1% de SDS três vezes à temperatura ambiente. A análise de sinais hibridizados foi realizada em um analisador de imagens BAS 2000 (Fuji Photo Film, Tóquio, Japão)
avaliação de Bibliotecas cDNA
Gel electroforese. A distribuição de tamanhos de inserção de cDNA foi examinada por electroforese de cRNAs sintetizadas por ARN polimerase T7 usando MB-AL e CDNAS plasmídicas MB-CL como modelos. Após a purificação usando um Mini Kit RNeasy, as cRNAs foram eletroforizadas em um gel de agarose contendo formaldeído com marcadores de RNA perfeitos (EMD Biosciences, Madison, WI, EUA). Depois de manchar o gel com SYBR® Green II (Invitrogen), a intensidade de coloração fluorescente foi analisada usando o software ImageQuant® (versão 5.0) em um FluorImager 595 (Biosciences Amersham).análise sequencial aleatória de passagem única. Sequências de 3 ‘ – End de clones de cDNA foram examinadas por sequenciação de DNA. DNA plasmídeo de 768 clones escolhidos aleatoriamente foram purificados usando um MFX-9600 Magnia® (Toyobo, Osaka, Japão) e analisados por um sequenciador RISA 384-capilar (Shimadzu, Kyoto, Japão) (16). Depois de aparar a sequência vetorial usando a Sequencher™ Software version 4.1 (Hitachi Software, Tóquio, Japão), As sequências cDNA obtidas com mais de 200 resíduos nucleotídeos foram agrupadas com entradas cDNA na Base de dados GenBank® e nossa base de dados interna do mouse. O exame da integridade das extremidades dos clones cDNA de 3′ foi realizado através de uma análise de se um hexâmero de sinal de poliadenilação canónico (5′-AATAAA-3’) ou as suas variantes de base única (11 hexâmeros de sinal) foram encontrados dentro dos 50 resíduos nucleótidos a montante da cauda poli(A) de cDNAs (21).microarray cDNA. Análises de Microarray foram realizadas para comparar populações de cDNA nas bibliotecas MB-AL e MB-CL. Microarrays de nylon caseiros com 3534 sondas foram preparados, e detecção radioisotópica foi realizada (22). Resumidamente, plasmídeos cDNA, que foram isolados no nosso instituto e para os quais sequências completas ou sequências finais já eram conhecidas, foram detectados utilizando GeneTAC™ RA1 (soluções genómicas, Ann Arbor, MI, EUA) na membrana de nylon de Biodyne B e, em seguida, fixados de acordo com as instruções do fabricante. A lista de cDNAs imobilizados neste microarray está disponível a pedido dos autores. Os cDNAs-alvo MB-AL e MB-CL foram preparados utilizando a transcrição reversa de cada uma das amostras de 1 µg cRNA utilizadas para a análise do tamanho da inserção, como descrito acima. Estas metas foram sintetizados e rotulada na mistura de reação contendo 1 µg de cada cRNA, 100 ng aleatório hexâmero, 1× first-strand buffer, 10 mM DTT, cada 800 µM dATP, dTTP, e dGTP, 800 nM, dCTP, 5 µL de dCTP (>2500 Ci/mmol; Amersham Biosciences), e 100 U SuperScript II RNase H – transcriptase reversa em temperatura ambiente por 10 min e, em seguida, a 42°C por 1 h. Após a lise alcalina e neutralização, o rotulado cDNAs foram purificadas com o QIAquick® PCR purification kit (QIAGEN) e contados. O cDNA-alvo foi sujeito a hibridação com o microarray de nylon cDNA na presença de 1 µg de ácido poliadenílico e 2,5 µg de Cot-1 DNA® do rato (Invitrogen) em 250 µL de tampão PerfectHyb™ (Toyobo) a 68°C durante a noite. Após uma lavagem rigorosa a 68 ° C com 2× SSC/1% SDS (duas lavagens de 15 minutos cada) e, em seguida, com 0,1× SSC/1% SDS (duas lavagens de 30 minutos cada), os sinais de hibridação foram detectados e analisados em um FLA-8000 (Foto de Fuji). Os pontos cDNA mostrando sinais mais fortes do que os do controle de fundo foram selecionados e submetidos a uma análise mais aprofundada. Após a realização da normalização global, foram obtidas parcelas de dispersão de intensidades de sinal de spots cDNA provenientes de alvos MB-AL e MB-CL.
Rna hibridização de blot. A hibridização RNA blot foi realizada para examinar os efeitos de viés de tamanho. As cRNAs MB-AL e MB-CL (cada 1, 5 µg, sintetizadas como descrito acima) foram electroforizadas em gel de agarose contendo formaldeído e transferidas para a membrana de nylon B de Biodyne. A sonda cDNAs foi preparada usando amplificação PCR. Os iniciadores foram concebidos com base nas sequências registadas na Base de dados GenBank ou na nossa base de dados interna.; os modelos eram os plasmídeos cDNA que tínhamos isolados. A sonda de ribossomal S6 proteína foi 724 bp em comprimento, amplificado com 5′-CGCTCGGCTGTGTCAAGATG-3′ e 5′-GAGGACAGCCTACGTCTCTTGG-3′ primers, e a detecção da proteína de choque térmico (hsp) 70, 940 bp de comprimento, foi amplificado com 5′-GATGGACAAGGCGCAGATCC-3′ e 5′-CTCGATGGTGGGTCCTGAGC-3′ primers. Estas sondas foram rotuladas com dCTP (aproximadamente 3000 Ci/mmol; Biociências de Amersham) usando o sistema de rotulagem de ADN RadPrime (Invitrogen). Após hibridação nocturna em tampão PerfectHyb a 65 ° C, As membranas foram lavadas sucessivamente com 0,1× SSC/1% SDS à temperatura ambiente durante 5 minutos e 15 minutos, e depois a 65°C durante 30 minutos. Os sinais de hibridação foram detectados em um analisador de imagem BAS 2000.
resultados e discussão
o método de construção da biblioteca cDNA que desenvolvemos neste estudo é mostrado na Figura 1 (os passos recentemente introduzidos são realçados com uma estrela). Este método consiste em duas rodadas de fase de amplificação cRNA, conversão de cRNAs em cDNAs, e clonagem recombinacional em um plasmídeo. O passo chave neste método é a ligação do adaptador para marcar o cDNA termina com a sequência Promotora SP6. Uma vez que possamos sintetizar com sucesso cDNAs neste formato, torna-se possível reverter especificamente a transcrição das cRNAs de primeira rodada que contêm a sequência Promotora SP6 no seu fim de 3′. As cRNAs de segunda rodada são sintetizadas com polimerase Rna SP6 usando as cDNAs de cadeia dupla resultantes como modelos. Após as duas rodadas de amplificação assistida por RNA polimerase, as cRNAs resultantes são então convertidas em cDNAs de cadeia dupla por transcrição reversa usando o attb2-Not-(dT)18 primer. O uso de attb2-Not-(dT)18 primer na transcrição reversa nos permite converter apenas os cRNAs carregando a seqüência 5′-(a)18GCGCGCCGC-3′ em suas extremidades 3′. Uma vez que estas modificações só devem permitir a amplificação e clonagem de cdnas que marcaram corretamente as extremidades, esperamos que este método irá minimizar o viés de tamanho durante a amplificação assistida por RNA polimerase.

para testar a eficácia do método, construímos bibliotecas cDNA por um método convencional (16) usando RNA total não amplificado do cérebro do rato (100 µg) e pelo método descrito acima usando 1 µg do RNA total do cérebro do rato. A tabela 2 resume as quantidades de cDNA e cRNA obtidas em cada etapa, calculadas como uma média de três séries experimentais independentes de construção MB-AL. Obtivemos uma média de 6,4×107 clones cDNA independentes como a saída final. Como este número de clones de cDNA deriva de um total de 36 ng cDNA gerado usando o protocolo de amplificação, calculamos que este método produz aproximadamente 1,2×1011 clones de cDNA a partir de 1 µg de RNA total.

Para examinar o tamanho do desvio de efeito durante a amplificação passos experimentalmente, foram comparadas as distribuições de tamanho do primeiro turno e o segundo turno cRNA por RNA blot hybridization análise. Nestes experimentos, o primeiro e o segundo round cRNAs foram hibridizados com SP6 UP primer e com não-(dT)18 oligonucleotídeo, respectivamente, porque estas sondas oligonucleotídicas detectam apenas cRNAs corretamente transcritas até o final. Como mostrado na Figura 2, o tamanho do RNA no pico do sinal de hibridação no cRNA de segunda rodada foi encontrado para ser quase o mesmo que no cRNA de primeira rodada, mas com uma ligeira mudança para tamanho menor. Nós suspeitamos que a truncação de cRNAs provavelmente ocorreu durante a síntese de cDNA segunda rodada com o SP6 UP primer.

a hibridação de ARN blot foi realizada para examinar o efeito de viés de tamanho sobre as cRNAs. Sinais de hibridação ao longo da direção da eletroforese de cRNA foram traçados. O tamanho do RNA foi determinado com base na mobilidade do marcador de escada de RNA. Linha azul, a cRNA primeira rodada hibridizada com sonda SP6 UP; linha vermelha, a cRNA segunda rodada hibridizada com sonda não-(dT)18; valor PSL, intensidade de sinal indicada arbitrariamente pelo software de Imagem (Foto de Fuji).
para avaliar mais a qualidade da biblioteca cDNA amplificada resultante, também comparamos várias características das bibliotecas cDNA geradas com e sem a amplificação (MB-AL e MB-CL, respectivamente). Examinamos primeiramente a distribuição de tamanho das inserções cDNA, como mostrado na figura 3A.os resultados dos sinais de intensidade de fluorescência obtidos a partir da imagem de coloração gel indicaram que os tamanhos de inserção cDNA de MB-AL e MB-CL eram semelhantes, mas o ponto de pico era ligeiramente menor em MB-AL (0,65 kb) em comparação com o MB-CL (0,8 kb). Em seguida, analisamos sequências de 3′-end de clones cDNA aleatoriamente amostrados de cada biblioteca. Os resultados de agrupamento para estas marcas de sequência expressas (ESTs) são mostrados na figura 3B e sugerem que a complexidade do MB-AL foi tão alta quanto a do MB-CL. No entanto, vale a pena notar que clones cDNA altamente redundantes desapareceram em MB-AL. Por uma análise estatística (teste Chi), demonstrou-se que a diferença de redundâncias entre MB-AL e MB-CL foi significativa (probabilidade de que os despedimentos em MB-AL e MB-CL são semelhantes é <0.001). Além disso, examinamos a integridade de 3 ‘ extremidades de cDNAs analisando se cada EST continha um hexâmero de sinal de poliadenilação ou não. Os resultados indicaram que 80,1% e 87,3% dos clones cDNA em MB-AL e MB-CL, respectivamente, continham sequências plausíveis de sinais de poliadenilação (21). A diminuição da taxa de ocorrência de hexâmeros de sinais de poliadenilação em MB-AL provavelmente indicou um aumento no número de clones de cDNA onde a síntese de cDNA foi preparada internamente devido a duas rodadas de preparação de dT.

MB-AL (mouse brain amplified cRNA-derived library) was avauated in comparison with a conventionally constructed library, MB-CL. A) comparação dos comprimentos de cDNA inseridos. As cRNAs transcritas in vitro foram fraccionadas em gel de agarose e manchadas. As suas intensidades fluorescentes ao longo da direcção da electroforese (indicadas como tamanho de ARN) são apresentadas em unidades fluorescentes relativas (rfus). B) resultados de agrupamento de sequências de fim de MB-AL e MB-CL 3′(ESTs). (C) Comparison of the cDNA population in MB-AL and MB-CL. Sinais obtidos por hibridização de microarray foram traçados. (D) resultados de hibridização de RNA blot sobre proteína S6 ribossomal e hsp 70. MB-AL cRNAs estavam na faixa da esquerda e as de MB-CL estavam na faixa da direita (indicadas por AL e CL, respectivamente). As pontas de flecha indicam as posições do tamanho previsto de RNA de cada gene. HSP, proteína de choque térmico.
por hibridização de microarray, examinamos a população cDNA de MB-AL em comparação com a de MB-CL. A figura 3C mostra um gráfico de dispersão de sinais de hibridação de alvos MB-AL e MB-CL. Os resultados indicaram uma correlação consideravelmente elevada de sinais de hibridação entre alvos MB-AL e MB-CL, sugerindo que não houve um efeito prejudicial da amplificação na população cDNA.
finalmente, realizamos a análise de hibridização de RNA blot para examinar os efeitos de viés de tamanho com atenção aos genes de limpeza (figura 3D). Para as proteínas S6 ribossomais e HSP 70, os tamanhos de cRNA tanto em MB-AL como MB-CL foram semelhantes e comparáveis aos esperados das suas sequências de nucleótidos relatadas. No entanto, o padrão de hibridação do HSP 70 cRNAs em MB-AL foi ligeiramente diferente do MB-CL, indicando que o efeito de viés de tamanho não foi completamente removido.
O método de construção da biblioteca cDNA descrito neste relatório foi concebido para minimizar os desvios de tamanho durante os passos de amplificação assistida por ARN polimerase. Tal RNA polimerase-assisted library construction method from a small RNA pool for comprehensive sequencing analysis has not been well studied, while cDNA / cRNA library construction methods for microarray hybridization have been actively pursued. Embora Lukyanov et al. (23) and Piao et al. (24) métodos relatados usando a amplificação PCR para construir uma biblioteca cDNA a partir de uma quantidade submicrograma de RNA total, a amplificação PCR intrinsecamente tem desvantagens como tamanho grave e viés populacional e baixa fidelidade da amplificação cDNA. Foi por isso que tentamos desenvolver um método de construção da biblioteca cDNA baseado na amplificação assistida por RNA polimerase neste estudo.para este fim, concebemos uma nova estratégia de amplificação assistida por RNA polimerase, como mostrado na Figura 1, porque os métodos de Eberwine et al. ( 5), Huang et al. (10), and Lin et al. (11) continuam a apresentar desvantagens na preparação de um cDNA contendo plasmídeos adequado para uma análise abrangente da estrutura genética. No primeiro, a síntese de cDNA após a amplificação de cRNA é feita com hexâmeros aleatórios, e isso faz com que os cDNAs amplificados inevitavelmente mais curtos do que os originais (5,7). Neste último, a introdução de uma cauda homopolimérica no fim da primeira linha de cDNA por desoxinucleotidil transferase terminal (TdT) ou vírus Moloney murine leukemia (MMLV) reverse transcriptase activity (10,11) faz sentido para minimizar o efeito de viés de tamanho durante a amplificação assistida por RNA polimerase como no nosso método. No entanto, sabe-se que o método homopolimérico causa a truncação de cDNAs devido à preparação inesperada da síntese de cDNA a partir de um trecho homopolimérico de sequências internas de ARN. Uma vez que o método de ligação-adaptador descrito neste estudo poderia fornecer uma sequência específica para a preparação do cDNA, poderíamos esperar reduzir a extensão da truncação do cDNA usando o nosso método.
em princípio, nosso método só deve amplificar as cDNAs flanqueadas por uma cauda intrínseca de poli(a) e a sequência do adaptador SP6, que foram geradas na síntese de cDNA de primeira rodada. Os resultados mostrados na Figura 2 e na figura 3A foram essencialmente consistentes com isso, embora a população cDNA na faixa menor de tamanho foi um pouco aumentada. Os resultados da análise de blot de RNA (figura 3D) também mostraram que o efeito de viés de tamanho poderia ser reduzido, mas não completamente removido, mesmo em nosso método. Isto deve-se provavelmente ao facto de os cDNAs truncados adicionados por uma cauda de poli(A) e a sequência Promotora SP6 terem sido gerados durante a conversão de cRNA em cDNA. Esta visão foi apoiada pelo fato de que a taxa de ocorrência das sequências de sinal de poliadenilação em MB-AL foi significativamente menor do que a observada na biblioteca cDNA convencional (21). A truncação de cDNAs pode ocorrer durante a síntese de cDNA devido à primagem interna do RNA com o primer de cauda dT e o primer SP6. Uma vez que os troços homopoliméricos ocorrem frequentemente em mRNAs eucarióticos, o método de tailing homopolimérico pode impor um viés de tamanho mais grave na amplificação CRNA múltipla redondo do que o nosso método de ligação adaptador -. Embora o aumento da temperatura de reação durante a transcrição reversa e a diminuição da concentração de primer poderia suprimir a iniciação interna aberrante na síntese de cDNA em certa medida, sabe-se que é difícil eliminar completamente isso atualmente. Assim, embora tenhamos realizado amplificação cRNA de duas voltas para demonstração do protocolo geral, na prática, recomendamos saltar a amplificação cRNA de segunda rodada quando cRNA suficiente pode ser obtido na reação de primeira rodada.considerando estes resultados, concluímos que o nosso novo método é útil para a construção da biblioteca cDNA a partir de uma pequena quantidade de RNA inicial. De fato, nós temos usado com sucesso e rotineiramente este método para a construção de bibliotecas cDNA quando a quantidade total de RNA é inferior a 1 µg (dados não mostrados). Uma vez que 1 µg de RNA total pode ser recuperado a partir de 105-106 células de mamíferos ou 1 mg de tecidos de mamíferos, as bibliotecas de cDNA podem ser facilmente construídas a partir de células fraccionadas por feitores de células ou Microdissecção usando o nosso método. Além disso, como calculamos que este método nos permite obter mais de 105 clones cDNA a partir de 1 pg RNA total, que está próximo da quantidade total de RNA em uma única célula, uma biblioteca cDNA derivada de uma única célula pode ser construída com base nesta amplificação cRNA assistida por adaptador-ligação.
agradecimentos
os autores estão gratos pela instrução da análise estatística do Dr. Kazuharu Misawa; divisão de plasmid DNAs do Dr. Hisashi Koga; e ajuda técnica da Sra. Akiko Ukigai-Ando e do Sr. Takashi Watanabe. O estudo foi financiado por um subsídio de Kazusa DNA do Instituto de Pesquisa de R. O., R. F. K., e O. O.
Interesses Concorrentes Instrução
Os autores declaram não interesses concorrentes.
- 1. Carninci, P., C. Kvam, A. Kitamura, T. Ohsumi, Y. Okazaki, M. Itoh, M. Kamiya, K. Shibata, et al.. 1996. Clonagem de cDNA de alta eficiência a tempo inteiro por caçador de CAP biotinilado. Genomics 37: 327-336.Crossref, Medline, CAS, Google Scholar
- 2. Ohara, O., T. Nagase, K. Ishikawa, D. Nakajima, M. Ohira, N. Seki, and N. Nomura. 1997. Construction and characterization of human brain cDNA libraries suitable for analysis of cDNA clones encoding relatively large proteins. DNA Res. 4: 53-59.Crossref, Medline, CAS, Google Scholar
- 3. Suzuki, Y., K. Yoshitomo-Nakagawa, K. Maruyama, A. Suyama, and S. Sugano. 1997. Construção e caracterização de uma biblioteca cDNA completa enriquecida em comprimento e com 5’de fim enriquecido. Gene 200: 1149-1156.Crossref, Google Scholar
- 4. Ohara, O. E G. Temple. 2001. Construção da biblioteca cDNA direccional assistida pela reacção de recombinação in vitro. Nucleic Acids Res. 29:E22.Crossref, Medline, CAS, Google Scholar
- 5. Eberwine, J., H. Yeh, K. Miyashiro, Y. Cao, S. Nair, R. Finnel, M. Zettel, and P. Coleman. 1992. Análise da expressão genética em neurónios vivos. Procedimento. Natl. Acad. Ciência. USA 89: 3010-3014.Crossref, Medline, CAS, Google Scholar
- 6. Wang, E., L. D. Miller, G. A. Ohnmacht, E. T. Liu, e F. M. Marincola. 2000. Amplificação mRNA de alta fidelidade para o perfil genético. Conversao. Biotechnol. 18:457–459.Crossref, Medline, CAS, Google Scholar
- 7. Baugh, L. R., A. A. Hill, E. L. Brown, C. P. Caçador. 2001. Analysis Quantitative of mRNA amplification by in vitro transcription. Nucleic Acids Res. 29:E29.Crossref, Medline, CAS, Google Scholar
- 8. Zhumabayeva, B., L. Diatchenko, A. Chenchik, and P. D. Siebert. 2001. Uso de cDNA SMART™ – gerado para estudos de expressão genética em múltiplos tumores humanos. BioTechniques 30: 158-163.Link, CAS, Google Scholar
- 9. Gomes, L., R., Silva, B. S. Stolf, E. B. Cristo, R. Hirata, Jr., F. A. Soares, L. Reis, E. J. Neves, et al.. 2003. Comparative analysis of amplified and nonamplified RNA for hybridization in cDNA microarray. Anal. Bioquímica. 321:244–251.Crossref, Medline, CAS, Google Scholar
- 10. Huang, J., T. Lin, D. Chang, S. Lin, and S. Ying. 2003. Bcl-2 truncado, um potencial marcador pré-metástico no cancro da próstata. Bioquímica. Biophys. Res. Commun. 306:912–917.Crossref, Medline, CAS, Google Scholar
- 11. Lin, S. L. E S. Y. Ying. 2003. mRNA / cDNA library construction using RNA-polymerase cycling reaction. Métodos Mol. Biol. 221:129–143.Medline, CAS, Google Scholar
- 12. Matz, M. V. 2003. Amplificação de piscinas cDNA representativas a partir de quantidades microscópicas de tecido animal. Métodos Mol. Biol. 221:103–116.Medline, CAS, Google Scholar
- 13. Petalidis, L., S. Bhattacharyya, G. A. Morris, V. P. Collins, T. C. Freeman, e P. A. Lião. 2003. Amplificação Global do mRNA por template-switching PCR: linearidade e aplicação à análise de microarray. Nucleic Acids Res. 22:e142.Crossref, Google Scholar
- 14. Polacek, D.C., A. G. Passerini, C. Shi, N. M. Francesco, E. Manduchi, G. R. Grant, S. Powell, H. Bischof et al.. 2003. Fidelity and enhanced sensitivity of differential transcription profiles following linear amplification of nanogram amounts of endothelial mRNA. Physiol. Genomics 13: 147-156.Crossref, Medline, CAS, Google Scholar
- 15. Wilson, C., S. D. Pepper, Y. Hey, E C. J. Miller. 2004. Os protocolos de amplificação introduzem erros sistemáticos mas reprodutíveis em estudos de expressão genética. BioTechniques 36: 498-506.Link, CAS, Google Scholar
- 16. Ohara, O., T. Nagase, G. Mitsui, H. Kohga, R. Kikuno, S. Hiraoka, Y. Takahashi, S. Kitajima, et al.. 2002. Caracterização da biblioteca cDNA fraccionada por tamanho gerada pelo método de recombinação in vitro assistido. DNA Res. 9: 47-57.Crossref, Medline, CAS, Google Scholar
- 17. Ohara, O. 2003. Construction of size-fractioned cDNA library assisted by an in vitro recombination reaction. Métodos Mol. Biol. 221:59–71.Medline, CAS, Google Scholar
- 18. Gubler, U. E B. J. Hoffman. 1983. Um método simples e muito eficiente para gerar bibliotecas cDNA. Gene 25: 263-269.Crossref, Medline, CAS, Google Scholar
- 19. Sambrook, J. and D. W. Russell. 2001. Preparação do ADN plasmídeo por lise alcalina com SDS, P. 1.31–1.42. Molecular Cloning, (3rd edition). CSH Laboratory Press, Cold Spring Harbor, NY.Google Scholar
- 20. Church, G. M. and W. Gilbert. 1984. Sequenciamento genómico. Procedimento. Natl. Acad. Ciência. USA 81: 1991-1995.Crossref, Medline, CAS, Google Scholar
- 21. Beaudoing, E., S. Freier, J. R. Wyatt, J. M. Claverie, and D. Gautheret. 2000. Padrões de Uso do sinal de poliadenilação variante em genes humanos. Genome Res. 10:1001-1010.Crossref, Medline, CAS, Google Scholar
- 22. Tanaka, T. S., S. A. Jaradat, M. K. Lopes, G. J. Kargul, X. Wang, M. J. Grahovac, S. Pantano, Y. Sano et al.. 2000. A expressão genoma do perfil da placenta e embrião de meia gestação usando um microarray cDNA de desenvolvimento de 15.000 ratos. Procedimento. Natl. Acad. Ciência. USA 97: 9127-9132.Crossref, Medline, Google Scholar
- 23. Lukyanov, K., L. Diatchenko, A. Chenchik, A. Nanisetti, P. Siebert, N. Usman, M. Matz, and S. Lukyanov. 1997. Construção de bibliotecas cDNA a partir de pequenas quantidades de RNA total usando o efeito supression PCR. Bioquímica. Biophys. Res. Commun. 230:285–288.Crossref, Medline, CAS, Google Scholar
- 24. Piao, Y., N. T. Ko, M. K. Lim, and M. Ko. 2001. Construction of long-transcripts enriched cDNA libraries from submicrogram amounts of total RNAs by a universal PCR amplification method. Genome Res. 11:1553-1558.Crossref, Medline, CAS, Google Scholar