
Why Develop Cap Analysis of Gene Expression?
Research by Piero Carninci and Yoshihide Hayashizaki in the late 1990s, which started with the cap trapper method, use of trehalose, the normalization/subtraction method, and a new cloning vector, set the stage for development of Cap Analysis of Gene Expression. Com o cap trapper, os híbridos cDNA/mRNA de comprimento completo são isolados, e o mRNA é biotinilado quimicamente na estrutura da tampa e as esferas magnéticas revestidas de streptavidina capturam os híbridos. Este avanço na série de grandes tecnologias de DNA é descrito pela natureza como o Marco 5 (www.nature.com/milestones/miledna/full/miledna05.html). seu objetivo no desenvolvimento de CAGE foi criar uma tecnologia para mapear de forma abrangente a grande maioria dos locais de início da transcrição humana e seus promotores. Na verdade, a tecnologia para traçar o perfil da actividade de transcrição genética em cada local promotor não existia até que CAGE chegou ao local. Messenger RNA (mRNA) representa uma ligação crítica entre a informação codificada em genes individuais em um genoma e a composição proteica que determina o destino de um organismo. Nós nos perguntamos: quais são as regiões genômicas, ou promotores, que impulsionam a expressão específica dos genes e suas RNAs singularmente diferentes? Na verdade, as coleções cDNA de comprimento total mostraram que a maioria dos genes tem mais de um local de partida de transcrição e, portanto, é bastante difícil identificar as regiões que controlam, ou seja, os promotores, responsáveis pela expressão das várias formas de transcrições.
Analisar a complexidade deste processo de transferência de informação, chamado de “transcrição”, requer o desenvolvimento de ferramentas moleculares sofisticadas que podem capturar os aspectos qualitativos e quantitativos da expressão genética. Assim, com o CAGE, a nossa transcrição original em todo o genoma, iniciando a tecnologia de detecção, podemos fazer perfis de expressão genética de alto nível com identificação simultânea dos locais de início transcriptional específicos do tecido/célula/condição (TSS), incluindo a análise de Utilização do promotor. A gaiola baseia-se na preparação e sequenciação de concatamers de marcas de ADN derivadas dos 20 nucleótidos iniciais de 5′ end mRNAs, que reflectem a concentração original de mRNA na amostra analisada (frequência de ARN).
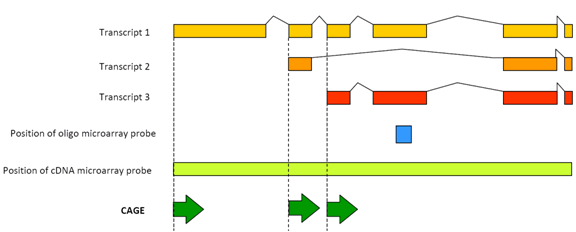
Figura 1: A gaiola detecta a actividade transcritiva de cada transcrição do promotor.as marcas sequenciais expressas (Tse) foram utilizadas em fases iniciais do desenvolvimento da tecnologia para identificar os elementos promotores, alinhando-os com o genoma humano. No entanto, este processo é muito caro devido aos custos de manipulação de cDNAs físicas e sequenciamento de Sanger. Uma maneira de superar esses problemas é usar tecnologias de tagging, que foram desenvolvidas para detectar transcrições com uma sensibilidade que é pelo menos uma ordem de magnitude maior do que a sequenciação EST, então exaustivamente identificar transcrições, identificar seus promotores e correlacioná-los com perfis de expressão, contando as tags como uma medida digital de expressão genética.estas sequências (também chamadas de “tags”) são então alinhadas às sequências do genoma por simples procedimentos computacionais (chamados BLAST) e contadas, o que dá uma medição da frequência da expressão de RNA. Como essas marcas de seqüência identificam os locais iniciais da transcrição de RNA, eles também identificam as seqüências do genoma perto desses locais iniciais. As regiões vizinhas são as regiões Promotoras centrais, que são sequências genômicas que fazem com que os genes sejam transcritos nas muitas condições diferentes que são encontradas em muitos organismos complexos, de ratos a humanos.os atributos de CAGE
CAGE têm grandes vantagens em comparação com técnicas clássicas de detecção de expressões baseadas em microarray. De fato, identificando o promotor que causa transcrição de RNA em cada fenômeno biológico, tecido, célula, etc. podemos identificar os elementos regulatórios de DNA que são específicos para cada fenômeno biológico, olhando para as sequências que estão nos promotores das isoformas de RNA que estão sendo expressas nas amostras analisadas. Promotores contêm sequências específicas, ou fator de transcrição sítios de ligação (TFBS), que são reconhecidos por sua vinculação proteínas chamadas fatores de transcrição (TF) e de promover, ou, alternativamente, de reprimir, de transcrição. Usando métodos computacionais, nossos pesquisadores analisam promotores com perfis de expressão semelhantes para suas TFBS e, em seguida, identificam os TFs responsáveis pela produção transcritional do genoma. Ao contar o número de tags de gaiola para cada promotor dentro de um gene, podemos agora determinar não só o nível de expressão de RNA (isto é uma detecção digital de frequência), mas, o que é importante, também a partir de qual dos vários promotores alternativos o RNA é transcrito.
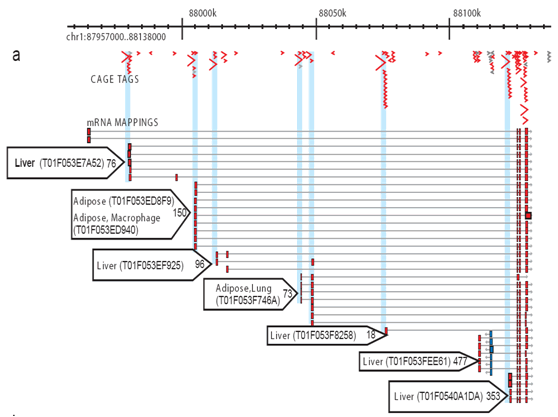
Figura 2: CAGE permite a caracterização completa das actividades em cada local promotor. Para cada biblioteca, são sequenciadas e alinhadas pelo genoma diversas marcas de gaiola, de modo a medir a actividade transcritional específica de cada promotor e a distinguir a contribuição de cada promotor. Este exemplo simplificado mostra a adipose e os promotores do núcleo do fígado. Setas azuis minúsculas: marcas individuais na gaiola; setas vermelhas: preferência pela utilização do promotor pelos tecidos; caixas vermelhas: regiões do promotor Central.
Como mencionado, a gaiola usa armadilhas como o primeiro passo para capturar as extremidades de 5′ das cDNAs, que são então transformadas em sequência curta (tags) de 20 a 27 nt, correspondendo ao mRNA TSSs,. Nós produzimos milhões de tags de gaiola para ratos e humanos usando tags de gaiola concatenadas com sequenciamento de Sanger, até que recentemente nos mudamos para a deepCAGE, para a qual usamos sequenciamento de segunda geração. até 2006, estávamos executando bibliotecas CAGE em nosso gasoduto sequenciador RISA original, que foi construído no final dos anos 90 e incluiu o único sequenciador capilar com uma série de 384 capilares que desenvolvemos em colaboração com a Shimadzu Corporation.
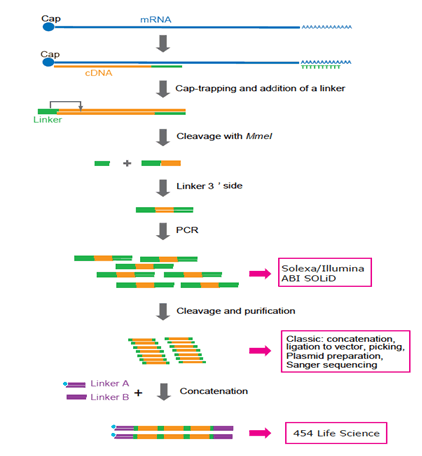
Figure 3: Representation of CAGE preparation protocol adapted to various platforms. Agora Solexa e ilumina são preferidos. 454 Ciências da Vida (sistema FLX) não é mais usado porque a concatenação requer ciclos PCR adicionais e manipulação complicada. No futuro, a tecnologia de sequenciamento de moléculas únicas será preferida porque a PCR pode não ser necessária.
embora a tecnologia de marcação e sequenciação tenha sido desenvolvida usando a análise serial do método de análise de genes (SAGE), CAGE é único porque é baseado no princípio de traçar o perfil do fim de 5′ de RNAs carregando um cap-site – todos os mRNAs e uma grande fração dos RNAs não codificantes. Desenvolvemos tags de gaiola longa de 27 nt para aumentar a eficiência do mapeamento.