
ważną częścią zrozumienia, jak te systemy działają w celu kontroli podziału komórek, było odkrycie, że p53 wpływa zarówno na raka, jak i nowotwór. Tyner i in. (2002) opracował strategię agenetyczną u myszy w celu porównania skutków braku p53 lub mniejszego niż normalne białko p53. Dwie transgeniczne linie myszy miały albo całkowitą delecję genu p53 (p53 -), albo skróconą postać p53 (p53m, mutant), która nie miała pierwszych sześciu eksonów genu p53 (fig. 2).
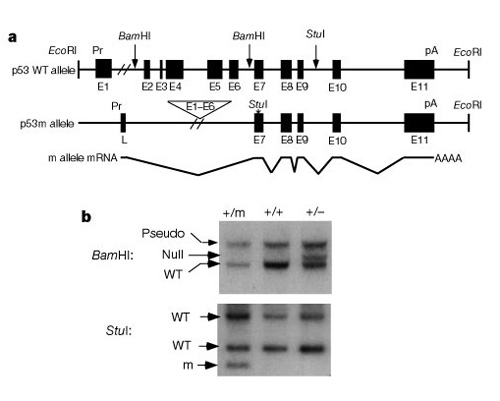
w pierwszej połowie badania porównano trzy grupy myszy: grupę 1, p53+/p53-(usunięcie jednej kopii p53); grupę 2, p53+/p53m(Mutant częściowej delecji) oraz grupę 3, p53+/p53+(Typ dziki, normalne). Tabela 1 przedstawia te trzy grupy i ich wyniki eksperymentalne związane z fenotypami raka i starzenia się. Co ciekawe, żadna z myszy z grupy 2, ze ściętym białkiem p53, nie rozwinęła guzów zagrażających życiu, podczas gdy 45% z grupy 3 (Typ dziki) i ponad 80% z grupy 1 rozwijało guzy zagrażające życiu. Myszy z grupy 2 miały również pośrednią długość życia pomiędzy bardzo krótką żywotnością myszy z grupy 1 i dłuższą żywotnością myszy z grupy 3. Wniosek z tych danych jest taki, że częściowa mutacja p53 zmniejszyła zachorowalność na raka, a jednocześnie wydawało się, że powoduje deficyt w długości życia, a nie wydłużony okres życia.
| Tabela 1. Experimental results from genetic mousestudies with p53 mutants | |||
| Genotype | Cancer phenotype | Agingphenotype | |
| Group 1 | p53+/p53- (complete deletion) | 80%had tumors | Muchshorter life span |
| Group 2 | p53+/p53m (partial mutant) | None | Shorterlife span |
| Group 3 | p53+/p53+ (wild type) | 45%had tumors | Normallife span |
| adaptacja z Tyner etal. 2002 | |||
autorzy stwierdzili również, że u myszy z grupy 2 rozwinęły się fenotypy charakterystyczne dla starych myszy, takie jak powolny odrost włosów i garbate kolce spowodowane zmianami szkieletowymi wcześniej niż myszy typu dzikiego (ryc. 3).

w drugiej połowie badania Tyner i koledzy zastanawiali się, czy Mutant p53 działa inaczej w obecności normalnego p53. Wyhodowali dodatkową transgeniczną linię myszy z mutantallelami p53-/p53m i odkryli, że myszy te nie mają silnej ochrony przed nowotworami i wykazują znacznie mniejszy efekt długości życia. Więc zmutowany p53 musiał jakoś współpracować z normalnym p53, aby uzyskać efekt. Ogólnie rzecz biorąc, badacze donoszą, że komórki z heterozygoty p53+/p53m okazały się około trzy razy trudniejsze do przetransformowania niż komórki typu dzikiego. Tak więc, chociaż komórki te były odporne na raka, ten p53+ / p53mbackground również spowodował wcześniejsze starzenie. Rzeczywiście, aktywność p53 w tych gatunkach wydaje się być znacznie wyższa niż jego aktywność w środowisku naturalnym type.It wydawało się, że taka zmiana, choć apriori, będzie dobra zarówno przeciwko rakowi, jak i starzeniu się, ale tak się nie stało.
później, Mooreet al. wykazano, że w komórkach kulturowych z tą samą mutacją powodującą obcięte białko p53, ta obcięta proteina weszła do jądra i kolokalizowała się z normalnym p53. Badali również okres półtrwania białka p53 w komórkach i odkryli, że heterozygoty z jedną kopią zmutowanego białka p53 miały około trzykrotnego wzrostu stabilności normalnego białka p53, w porównaniu z stabilnością samego dzikiego typu, co oznacza, że stabilność białka była poprawiona w stosunku do normy. Wyniki te w hodowanych komórkach wydłużyły się i potwierdziły badanie przeprowadzone przez Tynera i wsp. u myszy. Co więcej, był to pierwszy szlak odpowiedzi podziału komórek Odkryty do pracy przez p53 i wyraźnie modulować zarówno cancerincidence i starzenia.
w szlaku Rb, który może sygnalizować wyjście z cyklu podziału komórek, zdarzenia takie jak uszkodzenie DNA lub niewystarczająca replikacja prowadzące do krótkich telomerów na końcach chromosomów powodują zmniejszenie sygnalizacji CDK. To, inturn, zwiększa aktywność kinazy białkowej RB i w konsekwencji zwiększa aktywność czynnika transkrypcyjnego E2F. Jakie jest znaczenie E2F? Czynnik ten wiąże się z promotorami podjednostek polimerazy RNA i innymi białkami potrzebnymi do rozpoczęcia fazy S i pomaga w rozpoczęciu podziału komórek (Campisi 2003; Weinberg 1995). Wydaje się zatem, że zarówno szlak p53, jak i Szlak RB oddziałują na te same mechanizmy kontroli cyklu komórkowego.