
wprowadzenie inhibitora cyklu komórkowego
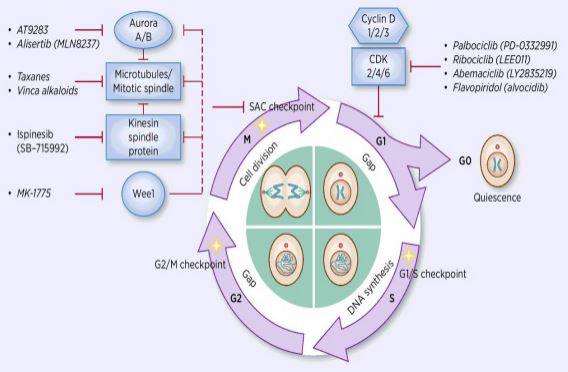
proces regulacji cyklu komórkowego polega na aktywacji lub inaktywacji różnych czynników regulacyjnych pod nadzorem punktów kontrolnych, inicjując w ten sposób proces replikacji DNA komórki i podziału na dwie komórki potomne. Wśród wielu regulatorów cyklu komórkowego kinaza zależna od cykliny (CDK) jest rdzeniem i jest to system sieciowy, który reguluje cykl komórkowy za pomocą cyklicznych i zależnych od cyklin inhibitorów kinazy (CKiS). CDK to klasa kinaz serynowo-treoninowych, a obecnie występuje 13 gatunków, w tym CDK1~13, które odgrywają rolę w regulacji cyklu komórkowego CDKs i transkrypcyjnej regulacji CDKs. Regulacja cyklu komórkowego jest w rzeczywistości regulacją punktów kontrolnych, przy czym najważniejsze są punkty regulacyjne G1 / s. Gdy cykl komórkowy jest stymulowany zewnętrznymi sygnałami, takimi jak czynniki wzrostu, podjednostka katalityczna CDK4/CDK6 wiąże się z Cyklindem podjednostki regulacyjnej, a reszty CDKs są aktywowane przez fosforylację/defosforylację. Po aktywacji CDK białko Rb ulega fosforylacji. Gen RB, znany również jako gen retinoblastoma, jest pierwszym sklonowanym genem supresorowym guza, a jego zdolność do tworzenia kompleksu z czynnikami transkrypcyjnymi (takimi jak E2F) po fosforylacji białka zostaje utracona. E2F odgrywa ważną rolę w regulacji cyklu komórkowego i indukuje ekspresję cykliny i CDK2 i tworzy kompleks Cyklina / CDK2, który dodatkowo fosforyluje białko RB i w pełni uwalnia E2F. następnie E2F wchodzi do jądra, aby aktywować serię cykli komórkowych do fazy S. W późnej fazie replikacji DNA podczas fazy s, CDK2 jest aktywowany przez cyklinę, która inaktywuje czynnik transkrypcyjny E2F w czasie, zapobiegając apoptozie spowodowanej przez uporczywie aktywowaną E2F. statystyki badań pokazują, że ponad 90% ludzkich nowotworów ma mutacje w powiązanych genach w ścieżkach CDK, Cyklina, cki i RB, przy czym CDK i odpowiadająca jej podjednostka regulacyjna Cyklina są najczęściej dysfunkcyjnymi. Ponadto, fluktuacje w cyklu komórkowym promują oporność na chemioterapię i zmniejszają skutki chemioterapii. Dlatego regulacja aktywności CDK / cyklin, która przywraca normalny cykl komórkowy, jest jedną ze strategii leczenia nowotworów.
inhibitory cyklu komórkowego są obecnie stosowane klinicznie
badacze leków skupili się na znalezieniu różnych typów inhibitorów CDK i cyklin jako najnowocześniejszych leków przeciwnowotworowych. Obecnie inhibitory CDK dzielą się głównie na endogenne i egzogenne. Największą klasą endogennych inhibitorów małocząsteczkowych są białka o niskiej masie cząsteczkowej, które są podzielone na dwie szerokie kategorie zgodnie z różnicami w funkcjach strukturalnych, a jedna klasa nazywa się dual specific family INK4, w tym p15, p16, p18, p19, która hamuje rodzinę białek. Białko hamujące kinazy związanej z Cyklindami wiąże się z odpowiednim wolnym CDK4, blokując w ten sposób Wiązanie CDK4 z odpowiednim cyklindem, tworząc katalityczny kompleks dimerów. Druga klasa nazywa się rodziną Kip, w tym P21, P27, P57. Ta rodzina białek może tworzyć trimer z kompleksem dimerycznym złożonym z cykliny E / CDK2 i cykliny / CDK1, blokując katalitycznie Aktywne Centrum dimeru. Hamowanie tych endogennych inhibitorów, w połączeniu z kompleksem kinaz, specyficznie reguluje jego aktywność, a tym samym precyzyjnie reguluje transformację komórki z fazy G1 do fazy S. Badania wykazały, że występowanie i rozwój wielu nowotworów są związane ze zmniejszoną ekspresją CDK/cyklin lub zmniejszoną ekspresją endogennych inhibitorów, takich jak delecja P16, która ma związek z rozwojem czerniaka, raka płuc, raka piersi i raka jelita grubego. Delecja białka P27 jest powszechna w raku piersi, raku prostaty, raku jelita grubego i raku przewodu pokarmowego. Dlatego delecja endogennego inhibitora CDKs lub mutacji genowej jest ważnym punktem odniesienia w diagnostyce nowotworu. Endogenne inhibitory małocząsteczkowe są również klasą ważnych niekodujących RNA odkrytych w ostatnich latach. Regiony miejsca docelowego wiążą się ze sobą, aby szybko i skutecznie degradować mRNA lub hamować translację białka, kontrolując białko na niższym lub optymalnym poziomie i wymagając aktywności życiowej. Odkryto ponad 10 mikroRNA biorących udział w regulacji cyklu komórkowego. Wśród nich miR1-2 i miR3-4 celują odpowiednio w CDK4, a cykl komórkowy jest zatrzymywany w fazie G1, która hamuje proliferację komórek nowotworowych; miR-22 celuje w komórki CDK6. Cykl jest stagnacja w fazie G1, która indukuje starzenie się w komórkach raka piersi. W różnych procesach biologicznych Mirna regulują przebieg cyklu komórkowego poprzez celowanie w E2F, CDK, Cyklinę, P21, P27, polimerazę DNA Alfa itd. promowanie lub blokowanie kluczowych regulatorów cyklu komórkowego. Do inhibitorów egzogennych należą antysensowne kwasy nukleinowe, przeciwciała, małe interferencyjne interferencje RNA (siRNA) i związki drobnocząsteczkowe. Związki drobnocząsteczkowe są najważniejszą klasą egzogennych inhibitorów CDK. W ostatnich latach, ponieważ zrozumienie struktury krystalicznej pozwala ludziom na prowadzenie badań symulacji molekularnej, dokonano przełomów w projektowaniu i rozwoju wysoce wydajnych i selektywnych badań nad chemicznymi inhibitorami CDK. Można powiedzieć, że takie związki mają nowych członków każdego dnia. Obecnie drobnocząsteczkowe inhibitory CDK można podzielić na następujące 13 kategorii: Roskowitinę i Ołomuniec, pirymidyny (PD-033299), flawonoidy (Flawopirydols), Tiazole (SNS03), antraceny i ich pochodne (SU951), piperydon (Paullones), imidazopirydynę, pirazolopirydynę (AZ703), pirazyny ( AT751), butyrolakton-1 (butyrolakton-1), scorpionine (UCN-01) i inne dwa gatunki. Trzynaście inhibitorów małocząsteczkowych weszło do badań klinicznych. Wszystkie są drobnocząsteczkowymi związkami heterocykli planarnych, które konkurują z ATP o wiązanie z miejscem wiązania ATP kinazy CDK. Eksperymenty in vivo wykazały, że CYC202 ma dobrą odporność na leki i dobrą fizjologiczną aktywność jamy ustnej i ma oczywiste działanie hamujące na guzy Lite U nagich myszy zaszczepionych ludzkim rakiem jelita grubego i komórkami raka macicy. W badaniach fazy Ib 10 pacjentów z rakiem jajnika przyjmowało CYC przez ponad 20 miesięcy, bez wzrostu liczby guzów lub ciężkich działań niepożądanych związanych z leczeniem, wśród których guz jednego pacjenta skurczył się o ponad 30%, a u niektórych pacjentów leczonych przez ponad rok stan jest stabilny. Badania kliniczne II Fazy wykazały, że sam CYKL202 ma nieco gorsze działanie i jest skuteczny w połączeniu z innymi lekami chemioterapeutycznymi. Trwają również badania kliniczne fazy IIb nad CYK202 w skojarzeniu z kapecytabiną w leczeniu raka piersi, w połączeniu z 2,2-difluorodeoksycytydyną lub cisplatyną w leczeniu raka płuc i raka nosogardzieli. Rozwój i zastosowanie małocząsteczkowej technologii interferencji RNA umożliwiły badanie ekspresji genu specyficznych cząsteczek docelowych interwencji, a wielu naukowców zaczęło interweniować w syntezę CDK / cykliny na poziomie genetycznym. Limaet al. transfekowano Sirna ukierunkowane na Cyklinę do Hep3B, HepG2, SNU449 (nadekspresja cykliny) i HuH7 (nadekspresja cykliny) i stwierdzono, że ekspresja cykliny została zmniejszona o 90% w komórkach. Synteza DNA jest znacznie zmniejszona, a komórki ulegają apoptozie. Galimberti et al. transfekowany siRNA ukierunkowany na Cyklinę, CDK2 i CDK1 do mysich komórek raka płuc, odpowiednio HOP-62, H-522 i H-23, i odkrył, że Cyklina/CDK2 może indukować apoptozę i hamować proliferację komórek raka płuc. Zmniejszona ekspresja CDK1 spowodowana interferencją siRNA cdk1 powoduje tylko zatrzymanie fazy komórki i spowalnia proliferację komórek; podczas gdy Ko-interferencja siRNA CDK1 i CDK2 prowadzi do jednoczesnego spadku ekspresji CDK1 i CDK2, powodując oporność w fazach cyklu komórkowego S I G2/M. Stagnacja wywołała również apoptozę komórek. Cao Yinfang i inne udane transfekcje rekombinowanego wektora ekspresyjnego CDK2/Sirna cykliny do komórek HepG2 wykazały, że ekspresja CDK2 i mRNA cykliny zmniejszyła się znacząco, cykl komórkowy został zatrzymany w fazie S, komórki fazy G1 znacznie wzrosły, aktywność kaspazy-3 zwiększyła się, komórki HepG2 przeszły apoptozę, a zmiany cyklu komórkowego były zgodne ze zmniejszoną proliferacją komórek HepG2 in vitro po transfekcji.
funkcja inhibitora cyklu komórkowego
wraz z pogłębieniem zrozumienia ważnej roli regulacji cyklu komórkowego w tworzeniu guza i apoptozie, regulacja cyklu komórkowego była dalej badana w oporności na chemioterapię nowotworu. Kinazy zależne od cyklin (CDK), które odgrywają rolę w napędzaniu silnika komórkowego podczas cyklu komórkowego, są idealnymi celami terapii nowotworowej. Większość komórek nowotworowych ma aktywację, nadekspresję genu cyklu podziału komórek (cdk) i wady funkcji CDKIs. Inhibitor CDK wywiera działanie przerwania, które hamuje cykl komórkowy. W ostatnich latach CDKIs stały się główną atrakcją terapii nowotworowej, która hamuje aktywność CDK w cyklu komórkowym. Seria badań klinicznych wykazała również, że pojedyncza aplikacja może mieć umiarkowane skutki. Jednak w połączeniu z tradycyjnymi cytotoksycznymi lekami chemioterapeutycznymi, CDKIs może znacznie zwiększyć działanie przeciwnowotworowe tradycyjnych leków chemioterapeutycznych. Dlatego badania nad przeciwnowotworowymi skutkami leków CDKIs i innych leków chemioterapeutycznych stały się gorącym punktem w obecnym leczeniu oporności na nowotwory.
Reference
- Bendris N, Lemmers B, Blanchard J M. Cell cycle, cytoszkielet dynamics and beyond: the many functions of cyclins and CDK inhibitors. Cykl Komórkowy. 2015, 14(12):1786-1798.
- Pitts T M, Davis S L, Eckhardt S G, et al. Celowanie w kinazy jądrowe w raku: rozwój inhibitorów kinazy cyklu komórkowego. Farmakologia & 2014, 142(2):258-269.
- Stone A, Sutherland R L, Musgrove E A. Inhibitors of cell cycle kinases: recent advances and future prospects as cancer therapeutics. Crit Rev Oncog. 2012, 17(2):175-198.
- Xu W, Mcarthur G. regulacja cyklu komórkowego i czerniak. Aktualne Raporty Onkologiczne. 2016, 18(6):34.
- Malumbres M, Barbacid M. Cell cycle, CDKs and cancer: a changing paradigm. Natura Ocenia Raka. 2009, 9(3):153-166.