
wprowadzenie
kompletne sekwencje genomu różnych organizmów, w tym ssaków, stały się ostatnio dostępne w wyniku szybkiego postępu w technologii sekwencjonowania DNA. Jednak, szczególnie u ssaków, analiza transkryptów nadal odgrywa kluczową rolę w wypełnianiu luki między genomem a proteomem. Dzieje się tak głównie dlatego, że obecnie nie możemy dokładnie przewidzieć struktur transkryptów pochodzących z konkretnego genu na podstawie samej informacji genomowej. Tak więc, jako metoda analizy transkryptów, Budowa biblioteki cDNA jest kluczowa, nawet w erze sekwencjonowania po genomie. Chociaż klonowanie cDNA interesujących genów przez PCR zapewniło uproszczoną alternatywną drogę do analizy struktur transkrypcji bez budowy biblioteki cDNA, konstrukcja biblioteki cDNA jest podejściem z wyboru, gdy duża liczba cDNA z jednego źródła mRNA ma być analizowana. Do tej pory włożono wiele wysiłku w opracowanie metody przygotowania wysokiej jakości bibliotek cDNA (1-4). Natomiast kwestia, w jaki sposób przygotować wysokiej jakości bibliotekę cDNA z małej puli RNA, nie została tak aktywnie rozwiązana. Stało się to jednak celem o wysokim priorytecie, ponieważ naukowcy często interesują się hipotetycznymi genami, które są przewidywane do ekspresji tylko w niektórych typach komórek lub tkanek w określonych warunkach, takich jak te obserwowane w próbkach patologicznych.
istnieje wiele doniesień opisujących użycie niewielkich ilości RNA źródłowego do wytwarzania amplifikowanego cDNA lub cRNA, z których można przygotować cele do analizy mikromacierzy (5-15). Ich ostatecznym celem jest uzyskanie pełnowymiarowego, nieuporządkowanego populacyjnie RNA, który jest wysoce reprezentatywny dla oryginalnego mRNA. Aby to zrobić, metody te zazwyczaj przyjmują PCR lub transkrypcję in vitro przez polimerazę RNA T7 w celu amplifikacji oryginalnego mRNA w postaci cDNA lub cRNA, odpowiednio. Chociaż porównanie profili ekspresji jest jednym z najbardziej efektywnych podejść do badania różnic w stanach fizjologicznych komórek lub tkanek, strukturalna charakterystyka transkryptów (np. identyfikacja alternatywnych wzorców splicingu, miejsca rozpoczęcia transkrypcji i miejsca zakończenia transkrypcji) nie może być wykonana za pomocą takiej analizy mikromacierzy. Analiza Sekwencyjna każdego transkryptu jest nieunikniona w tych przypadkach.
opracowaliśmy metodę, która pozwala na wykorzystanie niewielkiej ilości początkowego RNA do skonstruowania biblioteki cDNA odpowiedniej do klonowania genów i kompleksowej analizy sekwencjonowania. Metoda oznacza 3 'i 5′ końce cDNA pierwszej rundy odpowiednio sekwencjami promotora fagowego T7 i SP6, aby zminimalizować efekt odchylenia wielkości podczas amplifikacji. Po dwóch rundach amplifikacji cRNA, przekształciliśmy amplifikowane Crna w cDNA, sklonowaliśmy te produkty w plazmid, a następnie oceniliśmy otrzymaną bibliotekę cDNA w porównaniu z biblioteką zbudowaną konwencjonalną metodą (4,16,17).
materiały i metody
Amplifikacja cRNA
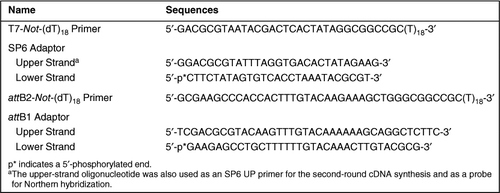
sekwencje oligonukleotydów użyte w tym badaniu przedstawiono w tabeli 1. Aby ustalić protokół, użyliśmy 1 µg całkowitego RNA przygotowanego z mózgu myszy ICR (8-tygodniowych samców myszy) jako szablonu. Mieszaninę (10 µL)1 µg całkowitego RNA i 100 pmol startera T7-Not-(DT)18 inkubowano w temperaturze 70°C przez 10 minut, a następnie schładzano na lodzie. Dwuniciową syntezę cDNA i ligację adaptera przeprowadzono zgodnie z protokołami systemu plazmidowego SUPERSCRIPT® (Invitrogen, Carlsbad, CA, USA) (4,16,17), z niewielkimi modyfikacjami. Krótko, do syntezy cDNA pierwszej nici dodano 4 µL buforu 5× pierwszej nici (Invitrogen), 1 µL 0,1 M ditiotreitolu (DTT), 1 µL 10 mM dNTPs, 1 µL (40 U) RNaseOUT™ (Invitrogen)i 1 µL wody do denaturowanej mieszaniny starterów RNA/T7-Not-(DT) 18 i inkubowano w 37°C przez 3 minuty w celu wyżarzania starterów. Następnie do mieszaniny reakcyjnej Dodano 2 µL (400 U) odwrotnej transkryptazy III RNazy H (Invitrogen) i temperaturę dostosowano do 50°C, aby rozpocząć syntezę cDNA pierwszej nici. Po 1 godzinie przeprowadzono syntezę cDNA drugiej nici, jak wcześniej opisali to Gubler i Hoffman (18); do mieszaniny cDNA pierwszej nici dodano 91 µL wody, 30 µL buforu 5× drugiej nici (Invitrogen), 3 µL 10 mM dNTPs, 1 µL (10 U) ligazy DNA Escherichia coli, 4 µL (40 U) polimerazy DNA E. coli i 1 µL (2 U) RNazy H E. coli i mieszaninę inkubowano w temperaturze pokojowej. 16°C przez 2 godziny.końce cDNA polerowano następnie polimerazą DNA 10 U T4 (Invitrogen) w temperaturze 16°C przez 5 minut, a reakcję zatrzymano przez dodanie 10 µl 0,5 m kwasu etylenodiaminotetraoctowego (EDTA). Otrzymane cDNA oczyszczono przez ekstrakcję fenolem / chloroformem / alkoholem izoamylowym (25:24:1), a następnie wytrącono Etanol, jak wcześniej opisano (17), z wyjątkiem użycia 1 µg Trna drożdży zamiast kwasu poliadenylowego jako nośnika. Wytrącony cDNA rozpuszczono w 50 µL TE (10 mM Tris-HCl, pH 8,0 i 1 mM EDTA), zmieszano z 30 µL 20% (w/v) glikolu polietylenowego (PEG) 6000/2, 5 M roztworu NaCl, a następnie inkubowano na lodzie przez 1 h (17). Po inkubacji mieszaninę cDNA odwirowywano w temperaturze 18 000× g w temperaturze 4°C przez 15 minut. Otrzymany osad cDNA przepłukano dwukrotnie 70% etanolem, wysuszono, a następnie zawieszono w 30 µL wody. Oczyszczone cDNA ligowano adapterem SP6 500 pmol (Tabela 1) stosując ligazę DNA 5 U T4 (Invitrogen) w 50-µL objętości reakcyjnej w temperaturze 16°C przez noc. Mieszaninę ligowaną adapterem rozcieńczono dwukrotnie wodą, a następnie oczyszczono cDNA za pomocą kolumny DNAclear™ (Ambion, Austin, TX, USA). Eluat (w 16 µL wody) zastosowano jako szablon do syntezy cRNA. Stosując zestaw MEGAscript® T7 (Ambion), Crna zsyntetyzowano w temperaturze 37°C przez noc w objętości reakcyjnej 40 µL. Po zdegradowaniu wzorcowych cDNA przez 4 U DNazy i, zsyntetyzowane Crna oczyszczono za pomocą zestawu RNeasy® Mini Kit (QIAGEN, Valencia, CA, USA) i eluowano 100 µL wody. Stężenie cRNA oznaczano za pomocą absorpcji w ultrafiolecie (UV). Do amplifikacji cRNA w drugiej rundzie użyto odpowiednio 2 µg zsyntetyzowanego Crna i 100 pmol startera SP6 UP (równego oligonukleotydowi oligonukleotydu górnego pasma adaptera SP6 przedstawionego w tabeli 1) jako szablon i starter. Drugą rundę syntezy cDNA przeprowadzono tak, jak w pierwotnej transkrypcji odwrotnej z źródłowego RNA, z tym wyjątkiem, że wyżarzanie starterowe SP6 UP przeprowadzono w temperaturze 50°C przez 3 min. Po oczyszczeniu otrzymanego dwuniciowego cDNA przez kolumnę DNAclear, 0,5 µg cDNA wykorzystano jako szablon do następnej amplifikacji cRNA wspomaganej polimerazą RNA SP6. Stosując zestaw MEGAscript SP6 (Ambion), syntezę cRNA prowadzono w temperaturze 37°C przez 6 godzin. po degradacji szablonu cDNA przy użyciu DNazy i, zsyntetyzowane Crna oczyszczono jak opisano powyżej.
konstrukcja Biblioteki cDNA
dwa mikrogramy powstałego Crna poddano dwuniciowej syntezie cDNA przy użyciu 100 pmol attb2-Not-(DT)18 primer (100 pmol attB2-Not – (DT) 18). tabela 1). Syntezę cDNA, stępienie końcowe, Oczyszczanie i ligację adaptera przeprowadzono tak, jak w poprzednich syntezach cDNA, z tym wyjątkiem, że adapter attB1 (Tabela 1, 500 pmol) ligowano do dwuniciowych cDNA. Po oczyszczeniu cDNA ligowanych adapterem attB1 przez kolejne zabiegi ekstrakcji fenolem/chloroformem/alkoholem izoamylowym, wytrącaniu etanolem i wytrącaniu PEG/NaCl w tej kolejności, rozpuszczono je w 50 µL TE i potraktowano Rnazą A (w końcowym stężeniu 10 µg/mL; Invitrogen) w celu degradacji zanieczyszczonego RNA w temperaturze 37°C przez 30 minut. Mieszaninę reakcyjną ponownie oczyszczono przez ekstrakcję fenolem / chloroformem / alkoholem izoamylowym, a następnie wytrącono etanol i wytrącono PEG / NaCl. Otrzymane cDNA rozpuszczono w 15 µL TE, a ich ilość oszacowano na podstawie intensywności barwienia fluorescencyjnego po elektroforezie na żelu agarozowym (4). CDNA ligowane attB1 (36 ng) poddano reakcji rekombinacji in vitro (System Gateway®; Invitrogen) z 250 ng wektorem dawcy attP-pSP73, jak opisano wcześniej (4,16,17). Krótko mówiąc, cDNA ligowane attB1 i Wektor dawcy attP-pSP73 zmieszano i inkubowano w 10-µL objętości reakcyjnej zawierającej 2 µL buforu reakcyjnego clonase™ 5× BP i 2 µL KLONAZY BP (obie z Invitrogenu) w ciągu nocy w temperaturze 25°C. Mieszaninę cDNA potraktowano 2 µg proteinazy K (Invitrogen) w temperaturze 37°C przez 10 minut w celu ugaszenia reakcji i ekstrahowano fenolem/chloroformem/alkoholem izoamylowym, a następnie wytrącono etanolem z 2 µg drożdżowego tRNA jako nośnika. Wytrącony cDNA rozpuszczono w 10 µL wody i 1 µL mieszaniny wykorzystano do transformacji komórek E. coli ElectroMAX™ DH10B™ (Invitrogen). Po miareczkowaniu otrzymanej biblioteki cDNA plazmidy cDNA odzyskiwano z około 1 000 000 Kolonii metodą alkalicznego siarczanu dodecylu sodu (SDS), jak opisano wcześniej (4,19). Powstałe klony cDNA miały postać plazmidu przenoszącego miejsca attL. Ponieważ plazmidy przenoszące miejsca attB są wymagane w niektórych zastosowaniach, plazmidy te przekształcono w plazmidy przenoszące miejsca attB, jak opisano wcześniej (16). Biblioteka cDNA została oznaczona jako MB-AL (mouse brain amplified cRNA-derived library).
w ten sam sposób, jak opisano dla MB-AL, skonstruowaliśmy bibliotekę cDNA z nieamplifikowanego RNA (100 µg całkowitego RNA mózgu myszy) i oznaczyliśmy ją jako MB-CL (biblioteka umozliwiająca umozliwienie mózgu myszy).
badanie wpływu odchylenia wielkości na amplifikowane Crna
w celu zbadania wpływu odchylenia wielkości na Crna, pierwszą rundę Crna transkrybowaną przez polimerazę RNA T7 i drugą rundę Crna transkrybowaną przez polimerazę RNA SP6 przeprowadzono na 1,0% żelu agarozowym (1 µg każdy), a następnie na błonie nylonowej (Nylonowa membrana Biodyne® B; PALL, East Hills, NY, USA). Hybrydyzowano je odpowiednio z oligonukleotydem Oligonukleotydowym znakowanym 32P SP6 UP (Tabela 1) I Not-(DT)18 (5′-GCGGCCGCTTTTTTTTTTTTTTTTT-3′). Dziesięć pikomoli każdej sondy oznaczono ATP (około 3000 Ci / mmol; Amersham Biosciences, Piscataway, NJ, USA) przy użyciu kinazy polinukleotydowej T4 (Takara, Kioto Japonia). Po nocnej hybrydyzacji w buforze GMC (20) w temperaturze 45°C, membrany te były intensywnie przemywane w 1× standardowym roztworze cytrynianu soli fizjologicznej (SSC)/1% roztworu SDS trzy razy w temperaturze pokojowej. Analizę hybrydyzowanych sygnałów przeprowadzono na analizatorze obrazu BAS 2000 (Fuji Photo Film, Tokio, Japonia)
ocena bibliotek cDNA
elektroforeza żelowa. Rozkład wielkości insertów cDNA badano elektroforezą Crna zsyntetyzowanych przez polimerazę RNA T7 przy użyciu NotI-trawionych MB-AL i MB-CL plazmidowych cDNA jako szablonów. Po oczyszczeniu za pomocą zestawu Rneasy Mini, Crna poddano elektroforezie na żelu agarozowym zawierającym formaldehyd z doskonałymi markerami RNA (EMD Biosciences, Madison, WI, USA). Po barwieniu żelu za pomocą SYBR® Green II (Invitrogen) analizowano intensywność barwienia fluorescencyjnego za pomocą oprogramowania ImageQuant® (Wersja 5.0) na FluorImager 595 (Amersham Biosciences).
Random single-pass sekwencing analysis. 3 ’ – końcowe sekwencje klonów cDNA zbadano przez sekwencjonowanie DNA. Plazmid DNA z 768 losowo wybranych klonów oczyszczono za pomocą MFX-9600 Magnia® (Toyobo, Osaka, Japonia) i przeanalizowano za pomocą sekwencera kapilarnego RISA 384 (Shimadzu, Kioto, Japonia) (16). Po przycięciu sekwencji wektorowej za pomocą oprogramowania Sequencher™ w wersji 4.1 (Hitachi Software, Tokio, Japonia), uzyskane sekwencje cDNA, które były dłuższe niż 200 reszt nukleotydowych, zostały zgrupowane z wpisami cDNA w bazie danych GenBank® i naszej własnej bazie danych myszy. Badanie integralności 3 'końców klonów cDNA przeprowadzono poprzez analizę tego, czy w obrębie 50 reszt nukleotydowych przed ogonem poli (a) cDNA (21) znaleziono kanoniczny heksamer sygnałowy poliadenylacji(5′-AATAAA-3′) lub jego warianty jedno zasadowe (11 heksamerów sygnałowych).
mikromacierze cDNA. Przeprowadzono analizy mikromacierzy w celu porównania populacji cDNA w bibliotekach MB-AL I MB-CL. Przygotowano domowe Nylonowe mikromacierze cDNA z 3534 sondami i przeprowadzono detekcję radioizotopową (22). Krótko mówiąc, plazmidy cDNA, które zostały wyizolowane w naszym instytucie i dla których znane były już pełne sekwencje lub sekwencje końcowe, zostały zauważone przy użyciu GeneTAC™ RA1 (Genomic Solutions, Ann Arbor, MI, USA) na nylonowej membranie Biodyne B, a następnie zamocowane zgodnie z instrukcjami producenta. Lista cDNA unieruchomionych na tej mikromacierzy jest dostępna na życzenie autorów. Docelowe cDNA MB-AL I MB-CL przygotowano stosując odwrotną transkrypcję z każdej z próbek 1 µg cRNA użytych do analizy wielkości Insertu, jak opisano powyżej. Cele te zsyntetyzowano i oznaczono w mieszaninie reakcyjnej zawierającej 1 µg każdego cRNA, 100 ng przypadkowego heksameru, 1× bufor pierwszej nici, 10 mM DTT, każdy 800 µM dATP, dTTP i dgtp, 800 nM dCTP, 5 µL dCTP (>2500 Ci/mmol; Amersham Biosciences) i 100 U SuperScript II RNase H-100 U odwrotna transkryptaza w temperaturze pokojowej przez 10 minut, a następnie w temperaturze 42°C przez 1 godzinę. po lizie zasadowej i neutralizacji znakowane cDNA oczyszczono za pomocą zestawu do oczyszczania qiaquick® PCR (QIAGEN) i policzono. Docelową cDNA poddano hybrydyzacji z nylonową mikromacierzą cDNA w obecności 1 µg kwasu poliadenylowego i 2,5 µg mysich Cot-1 DNA® (Invitrogen) w 250 µL buforu PerfectHyb™ (Toyobo) w temperaturze 68°C przez noc. Po rygorystycznym myciu w temperaturze 68°C 2× SSC/1% SDS (dwa mycia po 15 minut każde), a następnie 0,1× SSC/1% SDS (dwa mycia po 30 minut każde), sygnały hybrydyzacji wykryto i przeanalizowano na FLA-8000 (Fuji Photo Film). Wybrano i poddano dalszej analizie spoty cDNA wykazujące silniejsze sygnały niż te z kontroli tła. Po przeprowadzeniu globalnej normalizacji uzyskano wykresy rozproszone intensywności sygnału spotów cDNA pochodzących z celów MB-AL I MB-CL.
hybrydyzacja RNA blot. Hybrydyzację RNA blot przeprowadzono w celu zbadania efektu odchylenia wielkości. Crna MB-AL I MB-CL (każde 1,5 µg, zsyntetyzowane jak opisano powyżej)poddano elektroforezie na żelu agarozowym zawierającym formaldehyd i przeniesiono na membranę nylonową Biodyne B. Sondę cDNA przygotowano przy użyciu amplifikacji PCR. Startery zostały zaprojektowane w oparciu o sekwencje zarejestrowane w bazie GenBank lub w naszej własnej bazie danych; szablony były plazmidami cDNA, które wyizolowaliśmy. Sonda rybosomalnego białka S6 miała długość 724 bp, Amplifikowano za pomocą starterów 5′-CGCTCGGCGGTGTCAAGATG-3′ i 5′-GAGGACAGCCTACGTCTCTTGG-3′, a sonda białka szoku cieplnego (hsp) 70, 940 bp długości, Amplifikowano za pomocą starterów 5′-GATGGACAAGGCGCAGATCC-3′ i 5′-GAGGACAGCCTACGCGCAGATCC-3′ i 5 ’- ctcgatggtgggtcctgagc-3 ’ podkłady. Sondy te znakowano dCTP (około 3000 Ci / mmol; Amersham Biosciences) przy użyciu Radprime DNA Labeling System (Invitrogen). Po nocnej hybrydyzacji w buforze PerfectHyb w temperaturze 65°C, membrany przemyto kolejno 0,1× SSC/1% SDS w temperaturze pokojowej przez 5 minut i przez 15 minut, a następnie w temperaturze 65°C przez 30 minut. Sygnały hybrydyzacji wykryto w analizatorze obrazu BAS 2000.
wyniki i dyskusja
metoda budowy biblioteki cDNA, którą opracowaliśmy w tym badaniu, pokazana jest na rysunku 1 (nowo wprowadzone kroki są wyróżnione gwiazdką). Metoda ta składa się z dwóch rund etapu amplifikacji cRNA, konwersji Crna do cDNA i rekombinacyjnego klonowania do plazmidu. Kluczowym etapem w tej metodzie jest podwiązanie adaptera w celu oznaczenia końców cDNA sekwencją promotora SP6. Gdy uda nam się z powodzeniem zsyntetyzować cDNA w tym formacie, staje się możliwe specyficzne odwrotne transkrypcje pierwszych rund Crna, które zawierają sekwencję promotora SP6 na ich końcu 3′. Crna drugiej rundy są syntetyzowane z polimerazą RNA SP6 przy użyciu powstających dwuniciowych cDNA jako szablonów. Po dwóch rundach amplifikacji wspomaganej polimerazą RNA, powstałe Crna są następnie przekształcane do dwuniciowych cDNA przez odwrotną transkrypcję przy użyciu startera attB2-Not – (dT)18. Zastosowanie startera attB2-Not-(dT)18 w odwrotnej transkrypcji pozwala na konwersję tylko Crna niosących sekwencję 5′-(a)18GCGCGC-3′ na ich końcach 3′. Ponieważ te modyfikacje powinny umożliwiać tylko amplifikację i klonowanie cDNA, które mają prawidłowo oznaczone końce, spodziewamy się, że ta metoda zminimalizuje odchylenie rozmiaru podczas amplifikacji wspomaganej polimerazą RNA.

przedstawiono etapy wzmacniania cRNA wspomaganego przez adaptor-ligation-assisted oraz następującą konstrukcję biblioteki cDNA. Aby utrzymać amplifikowane rozmiary cDNA z wielkościami cDNA pierwszej rundy, wprowadziliśmy pewne modyfikacje w konwencjonalnych metodach wspomaganych polimerazą RNA (wyróżnionych gwiazdami). Niebieskie i czerwone linie wskazują odpowiednio cDNA i RNA. E. coli, Escherichia coli.
aby przetestować skuteczność tej metody, skonstruowaliśmy biblioteki cDNA metodą konwencjonalną (16) z wykorzystaniem nieamplifikowanego całkowitego RNA z mózgu myszy (100 µg) i metodą opisaną powyżej z wykorzystaniem 1 µg całkowitego RNA z mózgu myszy. W tabeli 2 podsumowano ilości cDNA i cRNA uzyskane w każdym etapie, obliczone jako średnia z trzech niezależnych prób konstrukcji MB-al. Otrzymaliśmy średnio 6,4×107 niezależnych klonów cDNA jako końcowy wynik. Ponieważ ta liczba klonów cDNA pochodzi z 36 ng cDNA wygenerowanych przy użyciu protokołu amplifikacji, obliczamy, że ta metoda daje około 1,2×1011 klonów cDNA z 1 µg całkowitego RNA.

aby zbadać efekt odchylenia wielkości podczas etapów amplifikacji eksperymentalnie, porównaliśmy rozkład wielkości pierwszej i drugiej rundy cRNA za pomocą analizy hybrydyzacji RNA blot. W tych eksperymentach, pierwsza i druga runda Crna zostały hybrydyzowane z starterem SP6 UP i odpowiednio z Oligonukleotydem Not – (dT)18, ponieważ te sondy oligonukleotydowe wykrywają tylko Crna poprawnie transkrybowane do końca. Jak pokazano na fig. 2, stwierdzono, że rozmiar RNA w szczycie sygnału hybrydyzacji w drugiej rundzie cRNA jest prawie taki sam jak w pierwszej rundzie cRNA, ale z niewielkim przesunięciem na mniejszy rozmiar. Podejrzewamy, że obcięcie Crna prawdopodobnie miało miejsce podczas drugiej rundy syntezy cDNA za pomocą startera SP6 UP.

przeprowadzono hybrydyzację RNA blot w celu zbadania wpływu odchylenia wielkości na Crna. Wykreślono sygnały hybrydyzacji wzdłuż kierunku elektroforezy cRNA. Wielkość RNA oznaczano na podstawie ruchomości markera drabinkowego RNA. Niebieska linia, pierwsza runda cRNA hybrydyzowana z sondą SP6 UP; czerwona linia, druga runda cRNA hybrydyzowana z sondą Not – (dT) 18; wartość PSL, intensywność sygnału wskazywana dowolnie przez oprogramowanie Image Gauge (Fuji Photo Film).
w celu dalszej oceny jakości uzyskanej amplifikowanej biblioteki cDNA porównaliśmy również różne funkcje bibliotek cDNA generowanych z i bez amplifikacji (odpowiednio MB-AL I MB-CL). Najpierw zbadaliśmy rozkład wielkości płytek cDNA, jak pokazano na fig. 3A. wyniki sygnałów intensywności fluorescencji uzyskanych z obrazu barwienia żelu wykazały, że rozmiary płytek cDNA MB-AL I MB-CL były podobne, ale punkt szczytowy był nieco mniejszy w MB-AL (0,65 kb) w porównaniu do punktu MB-CL (0,8 kb). Następnie przeanalizowaliśmy 3 ’ – końcowe sekwencje losowo pobranych klonów cDNA z każdej biblioteki. Wyniki grupowania dla tych wyrażonych znaczników sekwencji (est) przedstawiono na fig. 3B i sugerują, że złożoność MB-AL była tak wysoka jak MB-CL. Warto jednak zauważyć, że w MB-AL zniknęły wysoce zbędne klony cDNA. Analiza statystyczna (test Chi) wykazała, że różnica w redundancjach między MB-AL I MB-CL była znacząca (prawdopodobieństwo, że redundancje w MB-AL I MB-CL są podobne, wynosi <0,001). Ponadto zbadaliśmy integralność 3 ’ końców cDNA, analizując, czy każdy EST zawierał heksamer sygnału poliadenylacji, czy nie. Wyniki wykazały, że 80,1% i 87,3% klonów cDNA w MB-AL i MB-CL, odpowiednio, zawierało wiarygodne sekwencje sygnałowe poliadenylacji (21). Spadek częstości występowania heksamerów sygnału poliadenylacji w MB-AL prawdopodobnie wskazywał na wzrost liczby klonów cDNA, w których synteza cDNA była wewnętrznie zagruntowana z powodu dwóch rund gruntowania dT.

MB-AL (mouse brain amplified cRNA-derived library) oceniono w porównaniu z konwencjonalnie skonstruowaną biblioteką MB-CL. A) porównanie długości płytki cDNA. In vitro transkrybowane Crna były frakcjonowane na żelu agarozowym i barwione. Ich natężenie fluorescencji wzdłuż kierunku elektroforezy (wskazany jako rozmiar RNA) są pokazane we względnych jednostkach fluorescencyjnych (RFU). B) grupowanie wyników sekwencji MB-AL I MB-Cl 3 ’ – end (ESTs). C) porównanie populacji cDNA w MB-AL I MB-CL. Wykreślono sygnały uzyskane przez hybrydyzację mikromacierzy. (D) wyniki hybrydyzacji RNA blot na temat rybosomalnego białka S6 i hsp 70. MB-AL Crna były na lewym pasie, a te z MB-CL były na prawym pasie (oznaczone odpowiednio przez AL i CL). Groty strzał wskazują pozycje przewidywanej wielkości RNA każdego genu. hsp, białko szoku cieplnego.
poprzez hybrydyzację mikromacierzy zbadaliśmy populację cDNA MB-AL w porównaniu z populacją MB-CL. 3C przedstawia wykres rozproszony sygnałów hybrydyzacji celów MB-AL I MB-CL. Wyniki wykazały znacznie wysoką korelację sygnałów hybrydyzacji między celami MB-AL I MB-CL, co sugeruje, że nie było szkodliwego wpływu amplifikacji w populacji cDNA.
na koniec przeprowadziliśmy analizę hybrydyzacji RNA blot, aby zbadać efekty zmiany rozmiaru z uwzględnieniem genów domowych (Rysunek 3D). Dla rybosomalnego białka S6 i hsp 70, rozmiary cRNA zarówno w MB-AL, jak i MB-CL były podobne i porównywalne do tych, których oczekiwano po zgłaszanych sekwencjach nukleotydowych. Jednak wzór hybrydacji HSP 70 Crna w MB-AL był nieco inny niż w MB-CL, co wskazuje, że efekt odchylenia wielkości nie został całkowicie usunięty.
metoda budowy biblioteki cDNA opisana w niniejszym raporcie została opracowana w celu zminimalizowania błędów wielkości podczas etapów amplifikacji wspomaganych polimerazą RNA. Taka metoda budowy biblioteki wspomaganej polimerazą RNA z małej puli RNA do kompleksowej analizy sekwencjonowania nie została dobrze zbadana, podczas gdy metody budowy biblioteki cDNA/cRNA do hybrydyzacji mikromacierzy były aktywnie prowadzone. Chociaż Lukyanov et al. (23) oraz Piao et al. (24) zgłoszone metody wykorzystujące amplifikację PCR do skonstruowania biblioteki cDNA z submikrogramowej ilości całkowitego RNA, amplifikacja PCR z natury rzeczy ma wady, takie jak poważne odchylenie wielkości i populacji oraz niska wierność amplifikacji cDNA. Dlatego w tym badaniu próbowaliśmy opracować metodę budowy biblioteki cDNA opartą na amplifikacji wspomaganej polimerazą RNA.
w tym celu opracowaliśmy nową strategię amplifikacji wspomaganej polimerazą RNA, jak pokazano na fig. (5), Huang et al. (10), oraz Lin i wsp. (11) nadal posiadają wady wytwarzania cDNA zawierającego plazmid, odpowiednie do kompleksowej analizy struktury genów. W pierwszym przypadku synteza cDNA po amplifikacji cRNA odbywa się za pomocą losowych heksamerów, co sprawia, że amplifikowane cDNA są nieuchronnie krótsze niż oryginalne (5,7). W tym ostatnim, wprowadzenie homopolimerycznego ogona na końcu pierwszej nici cDNA przez końcową transferazę deoksynukleotydylową (TDT) lub odwrotną aktywność transkryptazy wirusa białaczki myszy Moloney (MMLV) (10,11) ma sens, aby zminimalizować efekt odchylenia rozmiaru podczas amplifikacji wspomaganej polimerazą RNA, jak w naszej metodzie. Wiadomo jednak, że homopolimeryczna metoda tailingu powoduje obcinanie cDNA z powodu nieoczekiwanego rozpoczęcia syntezy cDNA z homopolimerycznego odcinka wewnętrznych sekwencji RNA. Ponieważ metoda adaptor-ligation opisana w tym badaniu może dostarczyć specyficznej sekwencji cDNA priming, możemy spodziewać się zmniejszenia stopnia cDNA truncation przy użyciu naszej metody.
w zasadzie nasza metoda powinna tylko wzmacniać cDNA flankowane przez wewnętrzny ogon Poli(a) i sekwencję adaptera SP6, które zostały wygenerowane w pierwszej rundzie syntezy cDNA. Wyniki pokazane na fig. 2 i Fig. 3A były zasadniczo zgodne z tym, chociaż populacja cDNA w mniejszym zakresie wielkości była nieco zwiększona. Wyniki analizy blot RNA (Rysunek 3D) wykazały również, że efekt odchylenia wielkości można zmniejszyć, ale nie całkowicie usunąć nawet w naszej metodzie. Było to prawdopodobnie spowodowane tym, że obcięte cDNA dołączone przez ogon Poli(a) i Sekwencja promotora SP6 były generowane podczas konwersji cRNA do cDNA. Pogląd ten został poparty faktem, że częstość występowania sekwencji sygnałowych poliadenylacji w MB-AL była znacznie niższa niż obserwowana w konwencjonalnej bibliotece cDNA (21). Podczas syntezy cDNA może dojść do obcinania cDNA w wyniku wewnętrznego gruntowania RNA starterem dT i starterem SP6. Ponieważ rozciąganie homopolimeryczne często występuje w eukariotycznych mRNA, homopolimeryczna metoda tailingu może nałożyć poważniejsze odchylenie wielkości na wielokrotne wzmocnienie cRNA niż nasza metoda ligacji adaptera. Chociaż podniesienie temperatury reakcji podczas odwrotnej transkrypcji i zmniejszenie stężenia startera może do pewnego stopnia stłumić nieprawidłowe wewnętrzne gruntowanie w syntezie cDNA, obecnie wiadomo, że jest to trudne do całkowitego wyeliminowania. W związku z tym, chociaż przeprowadziliśmy dwie rundy amplifikacji cRNA w celu wykazania ogólnego Protokołu, w praktyce zalecamy pominięcie drugiej rundy amplifikacji cRNA, gdy w reakcji pierwszej rundy można uzyskać wystarczającą ilość cRNA.
biorąc pod uwagę te wyniki, wnioskujemy, że nasza nowa metoda jest przydatna do budowy biblioteki cDNA z niewielkiej ilości początkowego RNA. W rzeczywistości z powodzeniem i rutynowo stosowaliśmy tę metodę do budowy bibliotek cDNA, gdy ilość całkowitego RNA jest mniejsza niż 1 µg (dane nie pokazane). Ponieważ 1 µg całkowitego RNA można odzyskać ze 105-106 komórek ssaków lub 1 mg tkanek ssaków, biblioteki cDNA można łatwo zbudować z komórek frakcjonowanych przez sortowniki komórek lub mikrodysekcję za pomocą naszej metody. Ponadto, ponieważ obliczamy, że ta metoda pozwala nam uzyskać więcej niż 105 klonów cDNA z 1 PG całkowitego RNA, który jest zbliżony do ilości całkowitego RNA w pojedynczej komórce, biblioteka cDNA pochodząca z pojedynczej komórki może być skonstruowana w oparciu o wspomaganą przez adaptor ligację amplifikację cRNA.
podziękowania
autorzy są wdzięczni za instrukcje dotyczące analizy statystycznej od Dr Kazuharu Misawy; podział plazmidowych DNA od Dr Hisashi Koga; oraz pomoc techniczną od Pani Akiko Ukigai-Ando i Pana Takashi Watanabe. Badanie zostało wsparte Grantem Instytutu Badań DNA Kazusa na rzecz R. O., R. F. K. i O. o.
Oświadczenie o konkurencyjnych zainteresowaniach
autorzy nie deklarują konkurencyjnych zainteresowań.
- 1. Carninci, P., C. Kvam, A. Kitamura, T. Ohsumi, Y. Okazaki, M. Itoh, M. Kamiya, K. Shibata, et al.. 1996. Wysokowydajne klonowanie cDNA o Pełnej długości za pomocą biotynylowanego Cap trapera. Genomics 37:327-336.Crossref, Medline, CAS, Google Scholar
- 2. Ohara, O., T. Nagase, K. Ishikawa, D. Nakajima, M. Ohira, N. Seki, and n. Nomura. 1997. Budowa i charakterystyka bibliotek cDNA ludzkiego mózgu odpowiednich do analizy klonów cDNA kodujących stosunkowo duże białka. DNA Res. 4:53-59.Crossref, Medline, CAS, Google Scholar
- 3. Suzuki, Y., K. Yoshitomo-Nakagawa, K. Maruyama, A. Suyama, and S. Sugano. 1997. Budowa i charakterystyka biblioteki cDNA pełnej długości i 5′-endowej. Gene 200: 1149-1156.Crossref, Google Scholar
- 4. Ohara, o. i G. Temple. 2001. Directional cDNA library construction assisted by the in vitro recombination reaction. Kwasy nukleinowe Res.29:e22.Crossref, Medline, CAS, Google Scholar
- 5. Eberwine, J., H. Yeh, K. Miyashiro, Y. Cao, S. Nair, R. Finnel, M. Zettel, and p. Coleman. 1992. Analiza ekspresji genów w pojedynczych żywych neuronach. Proc. Natl. Acad. Sci. USA 89:3010-3014.Crossref, Medline, CAS, Google Scholar
- 6. Wang, E., L. D. Miller, G. A. Ohnmacht, E. T. Liu, and F. M. Marincola. 2000. Wysokiej jakości wzmocnienie mRNA do profilowania genów. Nat. Biotechnol. 18:457–459.Crossref, Medline, CAS, Google Scholar
- 7. Baugh, L. R., A. A. Hill, E. L. Brown i C. P. Hunter. 2001. Analiza ilościowa amplifikacji mRNA metodą transkrypcji in vitro. Kwasy nukleinowe Res.29:e29.Crossref, Medline, CAS, Google Scholar
- 8. Zhumabayeva, B., L. Diatchenko, A. Chenchik, and P. D. Siebert. 2001. Zastosowanie generowanego przez SMART™cDNA do badań ekspresji genów w wielu ludzkich guzach. BioTechniques 30:158-163.Link, CAS, Google Scholar
- 9. Gomes, L., R. Silva, B. S. Stolf, E. B. Cristo, R. Hirata, Jr., F. A. Soares, L. Reis, E. J. Neves, et al.. 2003. Analiza porównawcza amplifikowanego i nieamplifikowanego RNA do hybrydyzacji w mikromacierzy cDNA. Anal. Biochem. 321:244–251.Crossref, Medline, CAS, Google Scholar
- 10. Huang, J., T. Lin, D. Chang, S. Lin, and S. Ying. 2003. Obcięty Bcl-2, potencjalny marker pre-metastyczny w raku prostaty. Biochem. Biophys. Res.Commun. 306:912–917.Crossref, Medline, CAS, Google Scholar
- 11. Lin, S. L. i S. Y. Ying. 2003. Budowa biblioteki mRNA / cDNA z wykorzystaniem cyklicznej reakcji polimerazy RNA. Metody Mol. Biol. 221:129–143.Medline, CAS, Google Scholar
- 12. Matz, M. V. 2003. Amplifikacja reprezentatywnych pul cDNA z mikroskopijnych ilości tkanki zwierzęcej. Metody Mol. Biol. 221:103–116.Medline, CAS, Google Scholar
- 13. Petalidis, L., S. Bhattacharyya, G. A. Morris, V. P. Collins, T. C. Freeman, and P. A. Lyons. 2003. Globalne wzmocnienie mRNA przez zmianę szablonu PCR: liniowość i zastosowanie do analizy mikromacierzy. Kwasy nukleinowe Res. 22: e142.Crossref, Google Scholar
- 14. Polacek, D. C., A. G. Passerini, C. Shi, N. M. Francesco, E. Manduchi, G. R. Grant, S. Powell, H. Bischof, et al.. 2003. Wierność i zwiększona czułość różnicowych profili transkrypcyjnych po liniowym amplifikacji nanogramowych ilości mRNA śródbłonka. Physiol. Genomics 13:147-156.Crossref, Medline, CAS, Google Scholar
- 15. Wilson, C., S. D. Pepper, Y. Hey I C. J. Miller. 2004. Protokoły amplifikacji wprowadzają systematyczne, ale powtarzalne błędy do badań ekspresji genów. BioTechniques 36: 498-506.Link, CAS, Google Scholar
- 16. Ohara, O., T. Nagase, G. Mitsui, H. Kohga, R. Kikuno, S. Hiraoka, Y. Takahashi, S. Kitajima, et al.. 2002. Charakterystyka biblioteki cDNA frakcjonowanej wielkością generowanej metodą wspomaganą rekombinacją in vitro. DNA Res. 9: 47-57.Crossref, Medline, CAS, Google Scholar
- 17. Ohara, O. 2003. Budowa biblioteki cDNA frakcjonowanej wielkością wspomaganej reakcją rekombinacji in vitro. Metody Mol. Biol. 221:59–71.Medline, CAS, Google Scholar
- 18. Gubler, U. And B. J. Hoffman. 1983. Prosta i bardzo wydajna metoda generowania bibliotek cDNA. Gene 25:263-269.Crossref, Medline, CAS, Google Scholar
- 19. Sambrook, J. and D. W. Russell. 2001. Wytwarzanie plazmidu DNA przez zasadową lizę z SDS, str. 1.31–1.42. Klonowanie molekularne, (wydanie III). Csh Laboratory Press, Cold Spring Harbor, NY.Google Scholar
- 20. Church, G. M. I W. Gilbert. 1984. Sekwencjonowanie genomowe. Proc. Natl. Acad. Sci. USA 81:1991-1995.Crossref, Medline, CAS, Google Scholar
- 21. Beaudoing, E., S. Freier, J. R. Wyatt, J. M. Claverie, and D. Gautheret. 2000. Wzorce wariantowego wykorzystania sygnału poliadenylacji w ludzkich genach. Genome Res. 10: 1001-1010.Crossref, Medline, CAS, Google Scholar
- 22. Tanaka, T. S., S. A. Jaradat, M. K. Lim, G. J. Kargul, X. Wang,M. J. Grahovac, S. Pantano, Y. Sano, et al.. 2000. Profilowanie ekspresji genomu łożyska i zarodka w połowie ciąży przy użyciu 15 000 mysich mikromacierzy cDNA. Proc. Natl. Acad. Sci. USA 97:9127-9132.Crossref, Medline, Google Scholar
- 23. Lukyanov, K., L. Diatchenko, A. Chenchik, A. Nanisetti, P. Siebert, N. Usman, M. Matz, and S. Lukyanov. 1997. Budowa bibliotek cDNA z małych ilości całkowitego RNA z wykorzystaniem efektu supresji PCR. Biochem. Biophys. Res.Commun. 230:285–288.Crossref, Medline, CAS, Google Scholar
- 24. Piao, Y., N. T. Ko, M. K. Lim i M. Ko. 2001. Budowa długich transkryptów wzbogaconych bibliotek cDNA z submikrogramów całkowitych RNA za pomocą uniwersalnej metody amplifikacji PCR. Genome Res. 11: 1553-1558.Crossref, Medline, CAS, Google Scholar