
dlaczego warto rozwijać analizę Cap ekspresji genów ?
badania Piero Carninci i Yoshihide Hayashizaki pod koniec lat 90., które rozpoczęły się od metody cap trapper, zastosowania trehalozy, metody normalizacji / odejmowania i nowego wektora klonowania, ustawiły etap rozwoju analizy Cap ekspresji genów. W przypadku cap Traper, hybrydy cDNA/mRNA o Pełnej długości są izolowane, a mRNA jest chemicznie biotynylowane na strukturze cap, a kulki magnetyczne pokryte streptawidyną wychwytują hybrydy. Ten postęp w serii głównych technologii DNA jest opisany przez naturę jako kamień milowy 5 (www.nature.com/milestones/miledna/full/miledna05.html). ich celem w rozwoju CAGE było stworzenie technologii kompleksowego mapowania zdecydowanej większości ludzkich miejsc początkowych transkrypcji i ich promotorów. W rzeczywistości technologia profilowania aktywności transkrypcji genów w każdym miejscu promotora nie istniała, dopóki CAGE nie pojawił się na miejscu. Messenger RNA (mRNA) stanowi krytyczny związek między informacjami zakodowanymi w poszczególnych genach w genomie a składem białek, który decyduje o losie organizmu. Zadaliśmy sobie pytanie: Jakie są regiony genomowe lub promotory, które napędzają specyficzną ekspresję genów i ich wyjątkowo różne RNA? Rzeczywiście, pełnowymiarowe Kolekcje cDNA wykazały, że większość genów ma więcej niż jedno miejsce wyjściowe transkrypcji, a zatem dość trudno jest zidentyfikować regiony kontrolujące, czyli promotory, odpowiedzialne za ekspresję różnych form transkryptów.
Analiza złożoności tego procesu transferu informacji, zwanego „transkrypcją”, wymaga opracowania wyrafinowanych narzędzi molekularnych, które mogą uchwycić zarówno jakościowe, jak i ilościowe aspekty ekspresji genów. Dzięki CAGE, naszej oryginalnej technologii wykrywania transkrypcji w całym genomie, możemy wykonywać profilowanie ekspresji genów z jednoczesną identyfikacją miejsc początkowych transkrypcji specyficznych dla tkanki/komórki/stanu (TSS), w tym analizę wykorzystania promotora. CAGE opiera się na przygotowaniu i sekwencjonowaniu konkatamerów znaczników DNA pochodzących z początkowych 20 nukleotydów z 5′ końcowych mRNA, które odzwierciedlają pierwotne stężenie mRNA w analizowanej próbce (częstotliwość RNA).
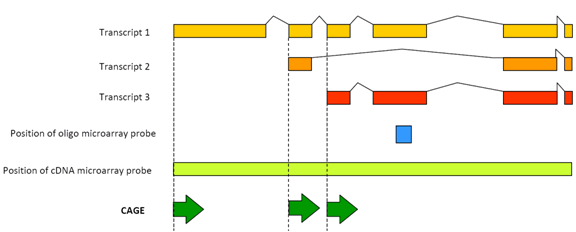
Rysunek 1: CAGE wykrywa aktywność transkrypcyjną każdego transkryptu promotora.
Expressed sequencing tags (est) były używane we wczesnych stadiach rozwoju technologii do identyfikacji elementów promotora poprzez dopasowanie ich do ludzkiego genomu. Jednak proces ten jest bardzo kosztowny ze względu na koszty obsługi fizycznych cDNA i sekwencjonowania Sangera. Jednym ze sposobów przezwyciężenia tych problemów jest użycie technologii znakowania, które zostały opracowane w celu wykrywania transkryptów z czułością, która jest co najmniej o jeden rząd wielkości większa niż sekwencjonowanie EST, a następnie wyczerpująco zidentyfikować transkrypty, zidentyfikować ich promotory i skorelować je z profilami ekspresji poprzez liczenie znaczników jako cyfrowej miary ekspresji genów.
sekwencje te (zwane również „tagami”) są następnie dopasowywane do sekwencji genomu za pomocą prostych procedur obliczeniowych (zwanych BLAST) I liczone, co daje pomiar częstotliwości ekspresji RNA. Ponieważ te znaczniki sekwencji identyfikują miejsca wyjściowe transkrypcji RNA, identyfikują one również sekwencje genomu w pobliżu tych miejsc wyjściowych. Sąsiednie regiony są głównymi regionami promotora, które są sekwencjami genomowymi, które powodują transkrypcję genów w wielu różnych warunkach, które występują w wielu złożonych organizmach, od myszy po ludzi.
atrybuty CAGE
Cage ma ogromne zalety w porównaniu do klasycznych technik wykrywania ekspresji opartych na mikromacierzy. W rzeczywistości, poprzez identyfikację promotora, który powoduje transkrypcję RNA w każdym zjawisku biologicznym, tkance, komórce itp. możemy zidentyfikować elementy regulacyjne DNA, które są specyficzne dla każdego zjawiska biologicznego, patrząc na sekwencje, które znajdują się w promotorach izoform RNA wyrażanych w analizowanych próbkach. Promotory zawierają specyficzne sekwencje lub miejsca wiążące czynnik transkrypcyjny (TFBS), które są rozpoznawane przez ich białka wiążące zwane czynnikami transkrypcyjnymi (TF) i promują lub alternatywnie hamują transkrypcję. Korzystając z metod obliczeniowych, nasi naukowcy analizują promotory o podobnych profilach ekspresji dla swoich TFB, a następnie identyfikują TFs odpowiedzialne za transkrypcyjną produkcję genomu. Zliczając liczbę tagów klatek dla każdego promotora w obrębie genu, możemy teraz określić nie tylko poziom ekspresji RNA (jest to Cyfrowe wykrywanie częstotliwości), ale, co ważne, także z którego z różnych alternatywnych promotorów RNA jest transkrybowany.
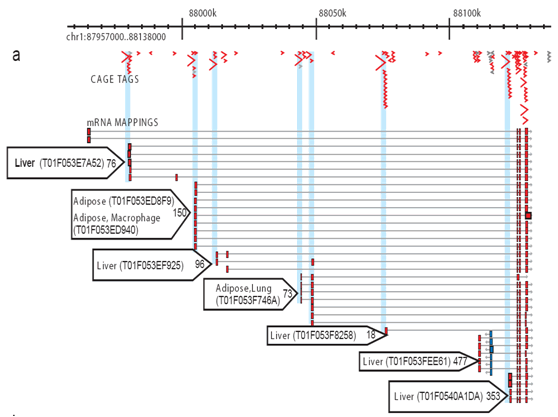
Rysunek 2: Klatka umożliwia kompleksowe profilowanie aktywacji w każdym miejscu promotora. Dla każdej biblioteki sekwencjonuje się i dopasowuje do genomu szereg znaczników klatek, dzięki czemu można zmierzyć specyficzną aktywność transkrypcyjną każdego promotora i rozróżnić wkład każdego promotora. Ten uproszczony przykład pokazuje promotory tkanki tłuszczowej i wątroby. Małe niebieskie strzałki: poszczególne znaczniki klatki; czerwone strzałki: preferencja wykorzystania promotora dla tkanek; czerwone pola: regiony promotora rdzenia.
jak wspomniano, CAGE wykorzystuje Cap-traping jako pierwszy krok do przechwytywania 5′ końców cDNA, które są następnie przekształcane w krótką sekwencję (znaczniki) od 20 do 27 nt odpowiadającą mRNA TSSs . Wyprodukowaliśmy miliony mysich i ludzkich znaczników klatek przy użyciu połączonych znaczników klatek z sekwencjonowaniem Sangera, aż niedawno przenieśliśmy się do deepCAGE, do którego używamy sekwencjonowania drugiej generacji.
do 2006 roku prowadziliśmy biblioteki klatek na naszym oryginalnym rurociągu sekwencjonowania RISA, który został skonstruowany pod koniec lat 90-tych i zawierał jedyny sekwencer kapilarny z tablicą 384 kapilar, który opracowaliśmy we współpracy z Shimadzu Corporation.
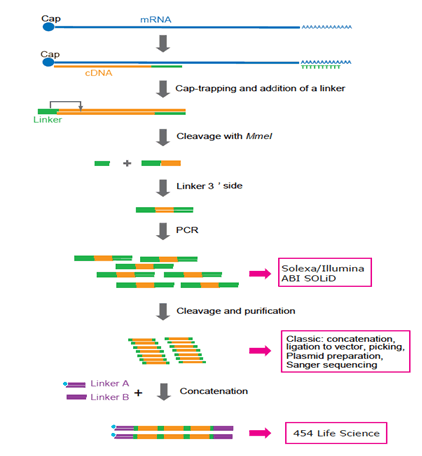
Rysunek 3: Przedstawienie protokołu przygotowania klatki dostosowanego do różnych platform. Teraz preferowane są Solexa i Illumina. 454 Life Sciences (system FLX) nie jest już stosowany, ponieważ konkatenacja wymaga dodatkowych cykli PCR i skomplikowanych manipulacji. W przyszłości technologia sekwencjonowania jednocząsteczkowego będzie preferowana, ponieważ PCR może nie być wymagane.
chociaż technologia tagowania i sekwencjonowania została opracowana przy użyciu metody seryjnej analizy analizy genów (SAGE),, CAGE jest unikalna, ponieważ opiera się na zasadzie profilowania 5′ końca RNA niosącego cap-site – all mRNA i dużą część niekodujących RNA. Opracowaliśmy 27-nt długie znaczniki klatkowe, aby zwiększyć wydajność mapowania.