
waarom Cap-analyse van genexpressie ontwikkelen?
onderzoek door Piero Carninci en Yoshihide Hayashizaki in de late jaren 1990, dat begon met de cap trapper methode, het gebruik van trehalose, de normalisatie/aftrekking methode, en een nieuwe klonen vector, zette het podium voor de ontwikkeling van Cap analyse van genexpressie. Met cap trapper worden cDNA/mRNA hybriden geïsoleerd, en mRNA is chemisch biotinylated op de cap structuur en streptavidin-gecoate magnetische parels vangen de hybriden. Deze vooruitgang in de reeks belangrijke DNA-technologieën wordt door de natuur beschreven als mijlpaal 5 (www.nature.com/milestones/miledna/full/miledna05.html hun doel in het ontwikkelen van kooi was om een technologie te creëren voor het volledig in kaart brengen van de overgrote meerderheid van menselijke transcriptie startende sites en hun promotors. In feite, bestond de technologie om de activiteit van gentranscriptie op elke promotorplaats te profileren niet totdat kooi op de scène kwam. BoodschappersRNA (mRNA) vertegenwoordigt een kritische verbinding tussen de informatie die in individuele genen op een genoom wordt gecodeerd en de eiwitmake-up die het lot van een organisme bepaalt. We vroegen ons af: wat zijn de genomische gebieden, of promotors, die de specifieke expressie van genen en hun uniek Verschillende RNAs aandrijven? Inderdaad, hebben de volledige verzamelingen cDNA aangetoond dat de meeste genen meer dan één plaats van het begin van de transcriptie hebben en zo is het vrij moeilijk om de controlegebieden te identificeren, dat is promotors, verantwoordelijk voor de uitdrukking van de diverse vormen van transcripten. het analyseren van de complexiteit van dit proces van informatieoverdracht, genaamd ’transcriptie’, vereist de ontwikkeling van geavanceerde moleculaire instrumenten die zowel de kwalitatieve als kwantitatieve aspecten van genexpressie kunnen vastleggen. Dus met CAGE, onze originele genoombrede transcriptie beginnende detectietechnologie, kunnen we high-through genexpressie profileren met gelijktijdige identificatie van de weefsel/cel/conditie specifieke transcriptionele startplaatsen (TSS), met inbegrip van promotorgebruik analyse. De kooi is gebaseerd op voorbereiding en het rangschikken van concatamers van de markeringen van DNA die uit de aanvankelijke 20 nucleotiden van eind mRNAs 5′ voortvloeien, die de originele concentratie van mRNA in de geanalyseerde steekproef (de frequentie van RNA) weerspiegelen.
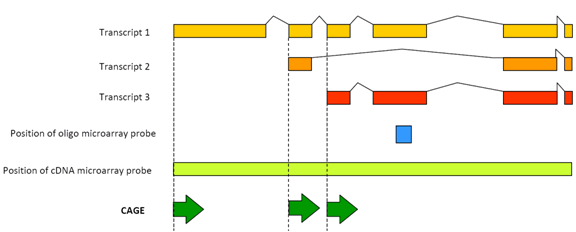
figuur 1: CAGE detecteert de transcriptionele activiteit van elke promotor transcript.
Expressed sequencing tags (ESTs) werden gebruikt in vroege stadia van de technologische ontwikkeling om promotorelementen te identificeren door ze op het menselijk genoom af te stemmen. Nochtans, is dit proces zeer duur wegens de kosten van het behandelen fysiek cDNAs en sanger rangschikken. Een manier om deze problemen te overwinnen is om het merken technologieën te gebruiken, die zijn ontwikkeld om transcripten met een gevoeligheid te ontdekken die minstens één orde van grootte groter is dan Est het rangschikken, dan uitputtend transcripten identificeren, hun promotors identificeren en hen met uitdrukkingsprofielen correleren door de markeringen als digitale maatregel van genuitdrukking te tellen.
Deze sequenties (ook wel “tags” genoemd) worden vervolgens uitgelijnd met genoomsequenties door eenvoudige computationele procedures (BLAST genoemd) en geteld, wat een meting van de frequentie van RNA-expressie geeft. Aangezien deze opeenvolgingsmarkeringen de beginplaatsen van de transcriptie van RNA identificeren, identificeren zij ook de genoomopeenvolgingen dicht bij die beginplaatsen. De naburige gebieden zijn de kerngebieden van de promotor, die genomic opeenvolgingen zijn die genen veroorzaken om bij de vele verschillende voorwaarden worden getranscribeerd die in vele complexe organismen, van muizen aan mensen worden ontmoet.
attributen van CAGE
CAGE heeft grote voordelen in vergelijking met klassieke microarray-gebaseerde expressiedetectietechnieken. In feite, door de promotor te identificeren die de transcriptie van RNA in elke biologische fenomenen, weefsel, cel enz. veroorzaken. wij kunnen de regelgevende elementen van DNA identificeren die specifiek voor elk biologisch fenomeen zijn door de opeenvolgingen te bekijken die in de promotors van de isovormen van RNA zijn die in de geanalyseerde steekproeven worden uitgedrukt. De promotors bevatten specifieke opeenvolgingen, of de plaatsen van de band van de transcriptiefactor (TFBS), die door hun bindende proteã NEN genoemd transcriptiefactoren (TF) worden erkend en, of alternatief, transcriptie bevorderen onderdrukken. Met behulp van computationele methoden, onze onderzoekers analyseren promotors met soortgelijke expressie profielen voor hun TFBS en dan identificeren de TFS verantwoordelijk voor de transcriptionele output van het genoom. Door het aantal KOOITAGS voor elke promotor binnen een gen te tellen, kunnen we nu niet alleen het RNA-expressieniveau bepalen (dit is een digitale detectie van frequentie) maar, belangrijker nog, ook van welke van de verschillende alternatieve promotors het RNA wordt getranscribeerd.
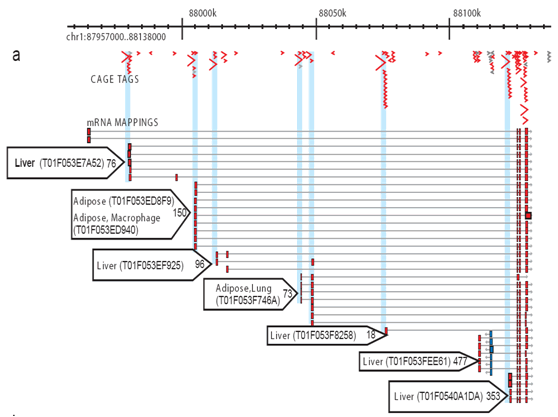
Figuur 2: CAGE maakt de uitgebreide profilering van activaten op elke promotorlocatie mogelijk. Voor elke bibliotheek, worden een aantal KOOIMARKERINGEN gerangschikt en afgestemd op het genoom zodat de specifieke transcriptional activiteit bij elke promotor kan worden gemeten en de bijdrage van elke promotor onderscheiden. Dit vereenvoudigde voorbeeld toont vet en lever kernpromotors. Tiny blue arrows: individuele kooi tags; rode pijlen: promotor gebruik voorkeur voor weefsels; rode dozen: Core promotor regio ‘ s.
zoals gezegd gebruikt CAGE cap-trapping als eerste stap om de 5′ uiteinden van de cDNA ‘ s te vangen, die vervolgens worden omgezet in een korte sequentie (tags) van 20 tot 27 nt die overeenkomt met de mRNA TSS, . We hebben miljoenen muis-en menselijke KOOITAGS geproduceerd met behulp van aaneengeschakelde KOOITAGS met Sanger-sequencing, tot we onlangs naar deepCAGE verhuisden, waarvoor we tweede generatie sequencing gebruiken. tot 2006 draaiden we CAGE libraries op onze originele RISA sequencing pipeline, die eind jaren 90 werd gebouwd en de enige capillaire sequencer bevatte met een reeks van 384 capillairen die we ontwikkelden in samenwerking met de Shimadzu Corporation.
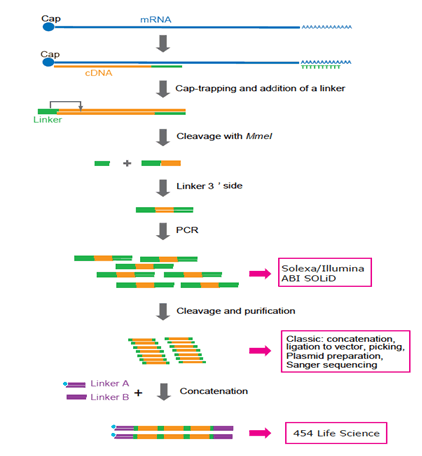
Figuur 3: weergave van het kooiprotocol aangepast aan verschillende platforms. Nu hebben Solexa en Illumina de voorkeur. 454 Life Sciences (FLX-systeem) wordt niet meer gebruikt omdat concatenation extra PCR-cycli en gecompliceerde manipulatie vereist. In de toekomst, zal het enig-molecuul rangschikken technologie de voorkeur hebben omdat PCR niet kan worden vereist.
hoewel de merking-en sequencingtechnologie is ontwikkeld met behulp van de seriële analyse van genanalyse (salie) – methode, is CAGE uniek omdat het gebaseerd is op het principe van het profileren van het 5′-uiteinde van RNAs met een cap-site-all mRNAs en een groot deel van de niet-coderende RNAs. We hebben 27-nt lange kooi tags ontwikkeld om de mapping efficiëntie te verhogen.