
Hvorfor Utvikle Cap Analyse Av Genuttrykk?
Forskning Av Piero Carninci og Yoshihide Hayashizaki på slutten av 1990-tallet, som startet med cap trapper-metoden, bruk av trehalose, normaliserings – /subtraksjonsmetoden og en ny kloningsvektor, satte scenen for utvikling Av Cap-Analyse av Genuttrykk. Med cap trapper, full-lengde cDNA/mRNA hybrider er isolert, og mRNA er kjemisk biotinylert på cap struktur og streptavidin-belagt magnetiske perler fange hybrider. Dette fremskrittet i serien av store DNA-teknologier er beskrevet Av Nature som Milestone 5 (www.nature.com/milestones/miledna/full/miledna05.html). deres formål med å utvikle CAGE var å skape en teknologi for omfattende kartlegging av det store flertallet av menneskelige transkripsjonsstartsteder og deres promotorer. Faktisk eksisterte ikke teknologien for å profilere aktiviteten til gentranskripsjon på hvert promotørsted før CAGE kom på scenen. Messenger RNA (mRNA) representerer en kritisk sammenheng mellom informasjonen kodet i individuelle gener på et genom og protein sminke som bestemmer en organismes skjebne. Vi spurte oss selv: hva er de genomiske regionene, eller promotørene, som driver det spesifikke uttrykket av gener og deres unikt forskjellige Rna? Faktisk har cDNA-samlinger i full lengde vist at de fleste gener har mer enn ett transkripsjonsstartsted, og det er derfor ganske vanskelig å identifisere de kontrollerende regionene, det vil si promotorer, som er ansvarlige for uttrykket av de ulike former for transkripsjoner. Å Analysere kompleksiteten i denne informasjonsoverføringsprosessen, kalt transkripsjon, krever utvikling av sofistikerte molekylære verktøy som kan fange både kvalitative og kvantitative aspekter av genuttrykk. SÅ MED CAGE, vår opprinnelige genom-wide transkripsjon starter deteksjon teknologi, kan vi gjøre høy hele genuttrykk profilering med samtidig identifisering av vev / celle / tilstand spesifikke transcriptional start sites (TSS), inkludert promoter bruk analyse. CAGE er basert på forberedelse OG sekvensering AV konkatamerer AV DNA-koder som stammer fra de første 20 nukleotidene fra 5 ‘ end mrna, som reflekterer den opprinnelige konsentrasjonen av mRNA i den analyserte prøven (RNA-frekvens).
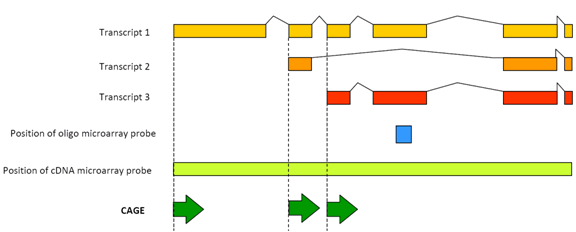
Figur 1: CAGE registrerer transkripsjonsaktiviteten til hver promotør-transkripsjon.Expressed sequencing tags (ESTs) ble brukt i tidlige stadier av teknologiutvikling for å identifisere promotorelementer ved å tilpasse dem til det menneskelige genomet. Denne prosessen er imidlertid svært dyr på grunn av kostnadene ved å håndtere fysiske cdnaer og Sanger-sekvensering. En måte å overvinne disse problemene på er å bruke taggingsteknologier, som er utviklet for å oppdage transkripsjoner med en følsomhet som er minst en størrelsesorden større ENN EST-sekvensering, deretter identifisere uttømmende transkripsjoner, identifisere deres promotorer og korrelere dem med uttrykksprofiler ved å telle kodene som et digitalt mål for genuttrykk.disse sekvensene (også kalt «tags») blir deretter justert til genomsekvenser ved enkle beregningsprosedyrer (kalt BLAST) og talt, noe som gir en måling av frekvensen AV RNA-uttrykk. Da disse sekvensmerkene identifiserer startstedene TIL rna-transkripsjonen, identifiserer de også genomsekvensene nær de startstedene. Naboregionene er kjernepromotor-regionene, som er genomiske sekvenser som forårsaker at gener blir transkribert ved de mange forskjellige forholdene som oppstår i mange komplekse organismer, fra mus til mennesker.
Attributter AV CAGE
CAGE har store fordeler sammenlignet med klassiske microarray-baserte uttrykksdeteksjonsteknikker. Faktisk ved å identifisere promotoren som forårsaker rna-transkripsjon i hvert biologisk fenomen, vev, celle etc. VI kan identifisere DNA-regulatoriske elementer som er spesifikke for hvert biologisk fenomen ved å se på sekvensene som er i promotorene AV rna-isoformene som uttrykkes i de analyserte prøvene. Promotorer inneholder spesifikke sekvenser, eller transkripsjonsfaktorbindingssteder (tfbs), som gjenkjennes av deres bindende proteiner kalt transkripsjonsfaktorer (tf) og fremmer, eller alternativt undertrykker, transkripsjon. Ved hjelp av beregningsmetoder analyserer våre forskere promotorer som har lignende uttrykksprofiler for DERES TFBS og identifiserer Deretter TFs som er ansvarlige for transkripsjonsutgangen av genomet. Ved å telle antall CAGE-koder for hver promotor i et gen, kan vi nå bestemme ikke bare rna-uttrykksnivået (dette er en digital deteksjon av frekvens), men viktigere, også fra hvilken av de forskjellige alternative promotorene RNA transkriberes.
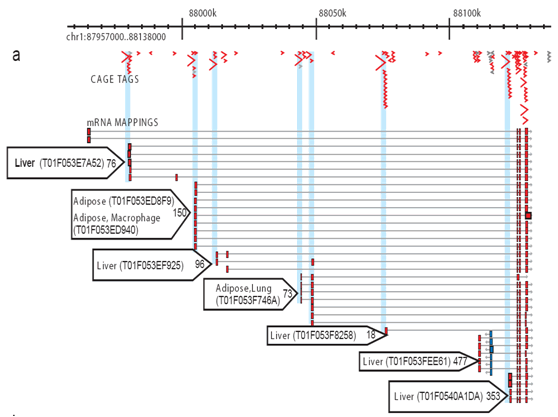
Figur 2: CAGE tillater omfattende profilering av aktiverer på hvert promotørnettsted. For hvert bibliotek er ET antall BURKODER sekvensert og justert til genomet, slik at den spesifikke transkripsjonelle aktiviteten hos hver promotør kan måles og bidraget fra hver promotør utmerker seg. Dette forenklede eksemplet viser adipose og lever kjerne promotorer. Små blå piler: individuelle BUR tags; røde piler: promoter bruk preferanse for vev; røde bokser: kjerne promoter regioner.
SOM nevnt, BRUKER CAGE cap-fangst som det første trinnet for å fange 5 ‘ endene av cDNAs, som deretter transformeres til kort sekvens (koder) av 20 til 27 nt tilsvarende mRNA TSSs, . Vi har produsert millioner av mus og menneskelige BURETIKETTER ved hjelp av sammenkoblede BURETIKETTER med Sanger-sekvensering, til vi nylig flyttet til deepCAGE, som vi bruker andre generasjons sekvensering. Frem til 2006 kjørte VI BURBIBLIOTEKER på VÅR opprinnelige RISA-sekvenseringsrørledning, som ble bygget på slutten av 90-tallet og inkluderte den eneste kapillærsekvenseren med en rekke 384 kapillærer som vi utviklet i samarbeid med Shimadzu Corporation.
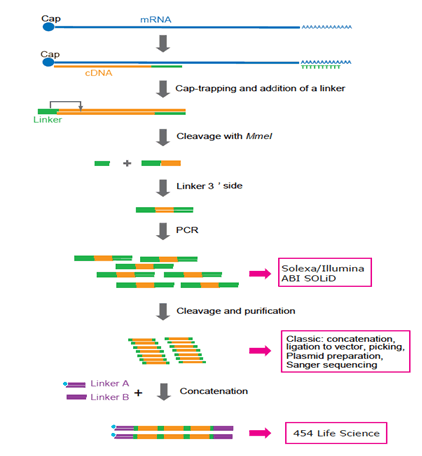
Figur 3: Representasjon AV BUR forberedelse protokoll tilpasset ulike plattformer. Nå Er Solexa og Illumina foretrukket. 454 Life Sciences (FLX system) brukes ikke lenger fordi sammenkobling krever ekstra PCR-sykluser og komplisert manipulasjon. I fremtiden vil enkeltmolekylsekvenseringsteknologi bli foretrukket fordi PCR kanskje ikke er nødvendig.Selv om tagging og sekvensering teknologi er utviklet ved hjelp av seriell analyse av genanalyse (SAGE) metode,, CAGE er unik fordi den er basert på prinsippet om profilering 5 ‘ enden Av Rna bærer en cap-site – alle mrna og en stor brøkdel av de ikke-kodende Rna. Vi har utviklet 27-nt lange BURETIKETTER for å øke kartleggingseffektiviteten.