
イントロダクション
哺乳類を含む様々な生物の完全なゲノム配列は、DNAシークエンシング技術の急速な進歩の結果として、最近利用可能になっている。 しかし、特に哺乳類では、転写物の分析は、まだゲノムとプロテオームの間のギャップを埋める上で重要な役割を果たしています。 これは、現在、ゲノム情報だけでは特定の遺伝子に由来する転写物の構造を正確に予測することができないためです。 したがって、転写物の分析のための方法として、cDNAライブラリーの構築は、ポストゲノム配列決定の時代であっても重要である。 PCRによる関心のある遺伝子のcDNAクローニングは、cDNAライブラリ構築なしで転写構造を分析するための単純化された代替ルートを提供しているが、cDNAラ 今日まで、高品質のcDNAライブラリーを調製するための方法の開発に多大な努力が払われてきた(1-4)。 対照的に、Rnaの小さなプールから高品質のcDNAライブラリを調製する方法の問題は、それほど積極的に対処されていません。 しかし、研究者は、病理学的サンプルで見られるような特定の条件下で特定の種類の細胞または組織でのみ発現すると予測される仮説的な遺伝子に
増幅されたcDNAまたはcRNAを生成するための少量のソースRNAの使用を記述する多くの報告があり、そこからマイクロアレイ分析のための標的を調製 それらの最終的な目標は、元のmRNAの高度に代表的な完全長の非集団バイアスRNAを得ることである。 これを行うために、これらの方法は、通常、PCRまたはT7RNAポリメラーゼによるin vitro転写を採用して、元のmRNAをcDNAまたはcRNAの形態でそれぞれ増幅する。 発現プロファイルの比較は、細胞や組織の生理学的状態の違いを調査するための最も効率的なアプローチの一つですが、転写物の構造特性(例えば、代替スプライシングパターン、転写開始部位、および転写終了部位の同定)は、このようなマイクロアレイ解析によって行うことはできません。 これらの場合、各転写物の配列解析は避けられません。
遺伝子クローニングや包括的なシーケンシング解析に適したcDNAライブラリーを構築するために、少量の出発RNAを使用できる方法を開発しました。 この方法は、増幅中のサイズバイアス効果を最小限に抑えるために、それぞれ、T7およびSP6ファージプロモーター配列と第一ラウンドcdnaの3’および5’端 二ラウンドcRNA増幅後、増幅したcrnaをcdnaに変換し、これらの生成物をプラスミドにクローニングし、得られたcDNAライブラリーを従来の方法(4,16,17)で構築したものと比較して評価した。
材料および方法
cRNA増幅
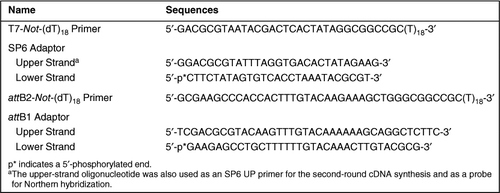
この研究で使用されたオリゴヌクレオチドの配列を表1に示す。 プロトコルを確立するために、我々はテンプレートとしてICRマウス脳(8週齢の雄マウス)から調製した1μ gの総RNAを使用しました。 1μ gの全RNAと1 0 0pmolのT7−Not−(d T)1 8プライマーの混合物(1 0μ l)を7 0℃で1 0分間インキュベートした後、氷上で急冷した。 二本鎖cDNA合成およびアダプター連結を、SUPERSCRIPT(登録商標)Plasmid System(Invitrogen,Carlsbad,C A,USA)(4,1 6,1 7)のプロトコルに従って、軽微な改変を加えて行った。 1Mジチオトレイトール(DT T)1μ l、1 0m M dNTPs1μ l、(4 0U)Rnaseout(商標)(Invitrogen)1μ l、および水1μ lを変性RNA/T7−Not−(D T)1 8プライマー混合物に添加し、3 7℃で3分間インキュベートして 次いで、反応混合物に2μ l(4 0 0U)SUPERSCRIPT III Rnase H−reverse transcriptase(Invitrogen)を添加し、温度を5 0℃に調整して第1鎖c DNA合成を開始した。 1時間後、GublerおよびHoffman(1 8)により以前に記載されたように第2鎖c DNA合成を行った;第1鎖c DNA混合物に、水9 1μ l、5×第2鎖緩衝液(Invitrogen)3 0μ l、1 0m M dNTPs3μ l、1μ l(1 0U)Escherichia coli DNA ligase、4μ l(4 0U)e.coli DNA polymerase、および1μ l(2U)e.coli Rnase Hを添加し、混合物を1 6℃で2時間インキュベートした。その後、cDNA末端を、1 0u t4dnaポリメラーゼ(Invitrogen)で1 6℃で5分間末端研磨し、1 0μ lの0. 得られたcDNAを、フェノール/クロロホルム/イソアミルアルコール(2 5:2 4:1)による抽出により精製し、続いて前述の(1 7)のようにエタノール沈殿させたが、ポリアデニル酸 沈殿したcDNAを、5 0μ lのTE(1 0m M Tris−Hcl、pH8. インキュベーション後、cDNA混合物を1 8,0 0 0×gで4℃で1 5分間遠心分離した。 得られたcDNAペレットを7 0%エタノールで2回すすぎ、乾燥させ、次いで3 0μ lの水に再懸濁した。 精製したcDNAを5 0 0pmol sp6アダプター(表1)で5U T4DNAリガーゼ(Invitrogen)を使用して、5 0μ lの反応体積中で1 6℃で一晩連結した。 アダプター連結された混合物を水で2倍に希釈し、次いで、Dnaclear(商標)カラム(Ambion,Austin,T X,USA)を用いてcDNAを精製した。 溶出液(水中1 6μ L)をcRNA合成のための鋳型として使用した。 Megascript(登録商標)T7キット(Ambion)を使用して、cRNAを3 7℃で4 0μ Lの反応容積で一晩合成した。 鋳型c DNAを4UのDnase iによって分解した後、合成したcRNAを、Rneasy(登録商標)Miniキット(QIAGEN,Valencia,C A,USA)を用いて精製し、1 0 0μ lの水で溶出した。 CRNAの濃度は、紫外線(UV)吸収によって決定した。 2回目のcrna増幅のために、2μ gの合成cRNAおよび1 0 0pmolのSP6upプライマー(表1に示されるsp6アダプターの上部鎖オリゴヌクレオチドに等しい)を鋳型およ 第二ラウンドのcDNA合成は、SP6upプライマーアニーリングを50℃で3分間行ったことを除いて、ソースRNAからの元の逆転写のように行った。 得られた二本鎖cDNAをDnaclearカラムで精製した後、0.5μ gのcDNAを、次のSP6RNAポリメラーゼ支援cRNA増幅のための鋳型として使用した。 Megascript SP6kit(Ambion)を用いて、cRNA合成を3 7℃、6時間行った。dnase iを用いて鋳型c DNAを分解した後、合成したcRNAを上記のようにして精製した。
cDNAライブラリー構築
得られたcrnaの二つのマイクログラムを、100pmol attb2-Not-(dT)18プライマーを用いた二本鎖cDNA合成に供した(表1)。 CDNA合成、末端鈍化、精製、およびアダプター連結工程は、ATTB1アダプター(表1、5 0 0pmol)が二本鎖cDNAに連結されたことを除いて、以前のcDNA合成と同様に実施した。 ATTB1アダプター連結cDNAを、フェノール/クロロホルム/イソアミルアルコール抽出、エタノール沈殿、およびPEG/Nacl沈殿の連続処理によりこの順序で精製した後、5 0μ lのTEに溶解し、Rnase A(最終濃度1 0μ g/ml;Invitrogen)で処理して、3 7℃で3 0分間、汚染RNAを分解した。 反応混合物を再びフェノール/クロロホルム/イソアミルアルコールによる抽出により精製し、続いてエタノール沈殿およびPEG/Nacl沈殿を行った。 得られたcdnaを15μ l TEに溶解し、その量をアガロースゲル上で電気泳動した後の蛍光染色強度から推定した(4)。 ATTB1連結cDNA(3 6ng)を、2 5 0ngのATTP−psp7 3ドナーベクターを用いたin vitro組換え反応(Gateway(登録商標)System;Invitrogen)に先に記載したように供した(4、1 6、1 7)。 要するに、ATTB1連結c DNAおよびATTP−psp7 3ドナーベクターを混合し、2μ lの5×BP Clonasetm反応緩衝液および2μ lのbp Clonase(両方ともInvitrogen製)を含む1 0μ lの反応容積中で、2 5℃で一晩 CDNA混合物を2μ gのプロテイナーゼK(Invitrogen)で37℃で10分間処理し、反応をクエンチし、フェノール/クロロホルム/イソアミルアルコールで抽出し、2μ gの酵母tRNAを担体としたエタノール沈殿を行った。 沈殿したcDNAを1 0μ lの水に溶解し、1μ lの混合物をElectromax(商標)D H1 0B(商標)e.coli細胞(Invitrogen)の形質転換のために使用した。 得られたcDNAライブラリーを滴定した後、cDNAプラスミドを、前述のようにアルカリドデシル硫酸ナトリウム(SDS)法により約1 0 0万コロニーから回収した(4,1 9)。 得られたcdnaクローンはattl部位を担持するプラスミドの形態であった。 いくつかの用途では、ATTB部位を有するプラスミドが必要とされるので、これらのプラスミドを、前述の(1 6)のようにATTB部位を有するプラスミドに変換した。 このcDNAライブラリーをMB−AL(mouse brain amplified cRNA−derived library)と命名した。MB-ALについて説明したのと同様に、非増幅RNA(100μ gのマウス脳トータルRNA)からcDNAライブラリーを構築し、MB-CL(マウス脳従来構築ライブラリー)と命名しました。
増幅されたcrnaに対するサイズバイアス効果の検査
crnaに対するサイズバイアス効果を調べるために、T7RNAポリメラーゼによって転写された第一ラウンドcrnaとSP6RNAポリメラーゼによって転写された第二ラウンドcrnaを1.0%アガロースゲル(各1μ g)上で実行し、ナイロン膜(Biodyne(登録商標)Bナイロン膜;PALL、East Hills、NY、USA)上でブロットした。 それらを、それぞれ、3 2P標識SP6upプライマー(表1)およびNot−(d T)1 8(5’−GCGGCCGCTTTTTTTTTTTTTTTTTTT−3’)オリゴヌクレオチドとハイブリダイズした。 各プローブの1 0ピコモルをATP(約3 0 0 0Ci/mmol)で標識した。; Amersham Biosciences,Piscataway,NJ,USA)を用いて、T4ポリヌクレオチドキナーゼ(Takara,Kyoto Japan)を用いた。 4 5℃のGMC緩衝液(2 0)中で一晩ハイブリダイゼーションした後、これらの膜を、1×標準クエン酸塩(SSC)/1%SDS溶液中で室温で3回広範囲に洗浄した。 ハイブリダイズされたシグナルの分析は、bas2000Image Analyzer(Fuji Photo Film,Tokyo,Japan)
cDNAライブラリーの評価
ゲル電気泳動上で行った。
ゲル電気泳動。
ゲル電気泳動。
ゲル電気泳動。 CDNA挿入サイズの分布は、テンプレートとしてNotI消化MB-ALとMB-CLプラスミドcdnaを使用してT7RNAポリメラーゼによって合成されたcrnaの電気泳動によって調 Rneasyミニキットを使用して精製した後、cRNAを、Perfect RNAマーカー(EMD Biosciences,Madison,WI,USA)を有するホルムアルデヒド含有アガロースゲル上で電気泳動した。 SYBR(登録商標)Green II(Invitrogen)でゲルを染色した後、蛍光染色強度を、Fluorimager5 9 5(Amersham Biosciences)上のImagequant(登録商標)(バージョン5.0)ソフトウェアを使用して分析した。
ランダムシングルパスシーケンシング解析。 CDNAクローンの3’末端配列は、DNA配列決定によって調べた。 無作為に選択した7 6 8クローンからのプラスミドDNAを、MFX−9 6 0 0Magnia(登録商標)(Toyobo,Osaka,Japan)を用いて精製し、RISA3 8 4−Capillary Sequencer(Shimadzu,Kyoto,Japan)によって分析した(1 6)。 1(Hitachi Software,Tokyo,Japan)を用いてベクター配列をトリミングした後、得られた2 0 0ヌクレオチド残基を超えるcDNA配列を、Genbank(登録商標)データベースおよび自社マウスデータベースに CDNAクローンの3’末端の整合性の検査は、正準ポリアデニル化シグナル六量体(5′-AATAAA-3’)またはその単一塩基変異体(11シグナル六量体)がcdnaのポリ(a)尾から上流50ヌクレオチド残基内に発見されたかどうかの分析によって行われた(21)。
cDNAマイクロアレイ。 マイクロアレイ分析は、MB-ALとMB-CLライブラリ内のcDNA集団を比較するために行われました。 3534プローブを備えた自家製cDNAナイロンマイクロアレイを調製し、放射性同位体検出を行った(22)。 簡単に説明すると、我々の研究所で単離され、完全配列または末端配列が既に知られていたcDNAプラスミドを、Genetac(商標)RA1(Genomic Solutions,Ann Arbor,MI,USA)を使用してBiodyne b nylon membrane上で発見し、次いで、製造者の指示に従って固定した。 このマイクロアレイ上に固定化されたcdnaのリストは、著者らからの要求に応じて入手可能である。 MB−A lおよびMB−CL標的c DNAを、上記のような挿入サイズ分析に使用した1μ gのcRNA試料のそれぞれからの逆転写を用いて調製した。 これらの標的を合成し、1μ gの各cRNA、1 0 0ngのランダム六量体、1×第1鎖緩衝液、1 0m MのDTT、各8 0 0μ MのDATP、DTTP、およびDGTP、8 0 0nMのdCTP、5μ LのdCTP(<div id=”1f0 6 7 6 8 8fb”></div>2 5 0 0Ci/mmol;Amersham Biosciences)、および1 0 0U Superscript I I Rnase H−アルカリ溶解および中和の後、標識したcdnaをqiaquick(登録商標)PCR PURIFICATION KIT(QIAGEN)で精製し、計数した。 5μ gのマウスCot−1DNA(登録商標)(Invitrogen)の存在下、2 5 0μ lのPerfecthyb(商標)緩衝液(Toyobo)中で、6 8℃で一晩、cDNAナイロンマイクロアレイとのハイブリダイゼーションを行った。 6 8℃で2×ssc/1%SDS(それぞれ1 5分間の2回の洗浄)、次いで、0.1×SSC/1%SDS(それぞれ3 0分間の2回の洗浄)で厳格に洗浄した後、ハイブリダイゼーション信号を検出し、FLA−8 0 0 0(Fuji Photo Film)で分析した。 背景対照のものよりも強いシグナルを示すcdnaスポットを選択し,さらなる分析を行った。 大域的正規化を行った後,MB-A lおよびMB-CL標的に由来するcdnaスポットのシグナル強度の散布図を得た。
RNAブロットハイブリダイゼーション。 サイズバイアス効果を調べるためにRNAブロットハイブリダイゼーションを行った。 MB−A lおよびMB−CLのcRNA(それぞれ1.5μ g、上記のように合成)をホルムアルデヒド含有アガロースゲル上で電気泳動し、Biodyne bナイロン膜に移した。 PCR増幅を用いてプローブcDNAを調製した。 プライマーはGenBankのデータベースか私達の社内データベースで登録されている順序に基づいて設計されていた; 鋳型は、我々が単離したcDNAプラスミドであった。 リボソームS6タンパク質のプローブは長さ724bpであり、5′-CGCTCGGCTGTGTCAAGATG-3’および5′-GAGGACAGCCTACGTCTCTTGG-3’プライマーで増幅され、熱ショックタンパク質(hsp)のプローブは長さ70、940bpで、5′-GATGGACAAGGCGCAGATCC-3’および5′-CTCGATGGTGGGTCCTGAGC-3’プライマーで増幅された。 これらのプローブを、Radprime DNA Labeling System(Invitrogen)を使用して、dCTP(約3 0 0 0Ci/mmol;Amersham Biosciences)で標識した。 1×SSC/1%SDSで、室温で5分間および1 5分間、次いで6 5℃で3 0分間連続的に洗浄した。 ハイブリダイゼーション信号は、BAS2000画像アナライザで検出されました。
結果と考察
本研究で開発したcDNAライブラリー構築法を図1に示します(新たに導入されたステップは星で強調表示されています)。 この方法は,crna増幅ステップ,cDNAへのcRNAの変換,およびプラスミドへの組換えクローニングの2ラウンドからなる。 この方法の重要なステップは、sp6プロモーター配列でcDNA末端をタグ付けするためのアダプター連結である。 この形式でcdnaを合成することができれば、SP6プロモーター配列を3’末端に含む最初のラウンドのcrnaを特異的に逆転写することが可能になる。 得られた二本鎖cDNAを鋳型として、SP6RNAポリメラーゼを用いて第2ラウンドcRNAを合成する。 2ラウンドのRNAポリメラーゼ支援増幅の後、得られたcRNAは、ATTB2−Not−(d T)1 8プライマーを用いた逆転写によって二本鎖cDNAに変換される。 逆転写におけるattb2-Not-(dT)18プライマーの使用は、配列5′-(A)18GCGGCCGC-3’をそれらの3’末端に運ぶcrnaのみを変換することを可能にする。 これらの修飾は、増幅と正しくタグ付けされた末端を持っているcdnaのクローニングを許可する必要がありますので、我々は、この方法は、RNAポリメラーゼ支援

アダプターライゲーション支援cRNA増幅および以下のcDNAライブラリ構築の手順を示します。 第一ラウンドのcdnaのものと増幅されたcDNAのサイズを維持するために、我々は、従来のRNAポリメラーゼ支援法(スターで強調)でいくつかの変更を導入しました。 青色および赤色の線は、それぞれcDNAおよびRNAを示す。 大腸菌(E.coli)、大腸菌(Escherichia coli)。
本方法の有効性を試験するために、マウス脳からの非増幅総RNA(100μ g)を用いた従来の方法(16)およびマウス脳からの総RNAの1μ gを用い 表2は、各工程で得られたcDNAおよびcRNAの量を要約し、MB−AL構築の3つの独立した実験実行の平均として計算した。 我々は、最終的な出力として6.4×107独立したcDNAクローンの平均を得た。 CDNAクローンのこの数は、増幅プロトコルを使用して生成された36ngのcDNAの合計に由来するので、我々は、この方法は、約1.2×1011のcDNAクローンを1μ gの総RNAか

増幅ステップ中のサイズバイアス効果を実験的に調べるために、我々は次のRNAブロットハイブリダイゼーション分析によって第一ラウンドと第二ラウンドcRNAのサイズ分布を比較した。 これらのオリゴヌクレオチドプローブは正しく最後に転写された唯一のcrnaを検出するため、これらの実験では、第一ラウンドと第二ラウンドcrnaは、そ 図2に示すように、第二ラウンドのcrnaのハイブリダイゼーションシグナルのピーク時のRNAサイズは、第一ラウンドのcRNAのそれとほぼ同じであるが、より小さ 我々は、crnaの切り捨ては、おそらくSP6アッププライマーと第二ラウンドcDNA合成中に行われたと思われる。

RNAブロットハイブリダイゼーションを行って、crnaに対するサイズバイアス効果を調べた。 Crna電気泳動の方向に沿ったハイブリダイゼーションシグナルをプロットした。 RNAサイズは、RNAラダーマーカーの移動度に基づいて決定した。 青い線、SP6UPプローブとハイブリダイズされた第一ラウンドのcRNA;赤い線、Not-(dT)18プローブとハイブリダイズされた第二ラウンドのcRNA;PSL値、画像ゲージソフトウェ得られた増幅されたcDNAライブラリーの品質をさらに評価するために、増幅の有無にかかわらず生成されたcDNAライブラリーの様々な特徴(それぞれMB-ALおよびMB-CL)を比較した。 ゲル染色画像から得られた蛍光強度シグナルの結果は、MB-ALとMB-CLのcDNAインサートのサイズは類似していたが、ピークポイントはmb-AL(0.65kb)でMB-CL(0.8kb)に比べてわずかに小さかったことを示した。 次に、我々は、各ライブラリからランダムにサンプリングされたcDNAクローンの3’末端配列を分析した。 これらの表現シーケンスタグ(ESTs)のクラスタリング結果を図3Bに示し、MB-ALの複雑さがMB-CLの複雑さと同じくらい高かったことを示唆しています。 しかし,mb-A lでは高度に冗長なcdnaクローンが消失したことは注目に値する。 統計分析(Chi検定)により、MB-ALとMB-CLの冗長性の差が有意であることが実証されました(MB-ALとMB-CLの冗長性が類似している確率は<0.001)。 さらに、我々は、各ESTがポリアデニル化シグナル六量体を含んでいるかどうかを分析することにより、cdnaの3’末端の整合性を調べた。 結果は、MB-ALとMB-CLのcDNAクローンの80.1%と87.3%は、それぞれ、もっともらしいポリアデニル化シグナル配列(21)が含まれていることを示した。 MB-A lにおけるポリアデニル化シグナルヘキサマーの発生率の減少は,dtプライミングの二ラウンドのためにcdna合成が内部プライミングされたcdnaクローンの数の増加を示した。

マイクロアレイハイブリダイゼーションにより、MB-ALのcDNA集団をMB-CLのcDNA集団と比較して調べました。 図3Cは、MB-ALターゲットとMB-CLターゲットのハイブリダイゼーション信号の散布図を示しています。 これらの結果は,MB-A lとM b-C L標的間のハイブリダイゼーションシグナルのかなり高い相関を示し,cdna集団における増幅の有害な影響はなかったことを示唆した。
最後に、ハウスキーピング遺伝子に注意してサイズバイアス効果を調べるためにRNAブロットハイブリダイゼーション解析を行いました(図3D)。 リボソームS6タンパク質とhsp70のために、mb-ALとMB-CLの両方のcRNAサイズは、それらの報告されたヌクレオチド配列から予想されるものと同様であった。 しかし、MB-ALにおけるhsp70crnaのハイブリダイゼーションパターンは、サイズバイアス効果が完全に除去されなかったことを示す、MB-CLのそれとはわずかに異
本報告書に記載されているcDNAライブラリー構築の方法は、RNAポリメラーゼによって支援された増幅ステップ中のサイズバイアスを最小限に抑えるた マイクロアレイハイブリダイゼーションのためのcDNA/cRNAライブラリ構築法が積極的に追求されている間、包括的な配列決定分析のための小さなRNAプール Lukyanovらが。 (2 3)およびPiao e t a l. (24)PCR増幅を用いた報告された方法全RNAのサブミクロン量からcDNAライブラリーを構築するためには、PCR増幅は本質的に、深刻なサイズおよび集団バイア そこで本研究では,RNAポリメラーゼ支援増幅に基づくcdnaライブラリー構築法の開発を試みた。この目的のために、図1に示すような新しいRNAポリメラーゼ支援増幅戦略を考案したのは、Eberwine et al. (5),Huang et al. (1 0)、およびLin e t a l. (11)包括的な遺伝子構造解析に適したプラスミド含有cDNAの調製には依然として欠点がある。 前者では、cRNA増幅後のcDNA合成はランダムなヘキサマーで行われ、これにより増幅されたcDNAは必然的に元のものよりも短くなります(5,7)。 後者では、末端デオキシヌクレオチジルトランスフェラーゼ(TdT)またはモロニーマウス白血病ウイルス(MMLV)逆転写酵素活性(10,11)による第一鎖cDNA末端にホモポリマー しかし、ホモポリマーテーリング法は、内部RNA配列のホモポリマーストレッチからのcDNA合成の予期せぬプライミングによるcdnaの切断を引き起こすことが知られている。 本研究で記載されているアダプターライゲーション法は、cDNAプライミングのための特定の配列を提供することができるので、我々は我々の方法を使用してcDNA
原理的には、我々の方法は、最初のラウンドcDNA合成で生成された固有のポリ(A)テールとSP6アダプター配列によって隣接するcdnaを増幅する必要があります。
図2および図3Aに示された結果は、これと本質的に一致していたが、より小さいサイズ範囲のcDNA集団は少し増加した。 RNAブロット解析の結果(図3D)はまた、サイズバイアス効果は減少することができるが、我々の方法でも完全に除去することはできないことを示した。 これはおそらく、ポリ(A)尾部およびSP6プロモーター配列によって付加された切り捨てられたcdnaがcrnaのcDNAへの変換中に生成されたためであった。 この見解は、MB-ALにおけるポリアデニル化シグナル配列の発生率が従来のcDNAライブラリー(21)で観察されたものよりも有意に低かったという事実によっ Cdnaの切り捨ては、dT尾プライマーとSP6プライマーとRNAの内部プライミングのためにcDNA合成中に発生する可能性があります。 ホモポリマーストレッチは真核生物のmrnaで頻繁に発生するため、ホモポリマーテーリング法は、我々のアダプターライゲーション法よりも複数ラウンドcRNA増幅に 逆転写中の反応温度を上昇させ,プライマー濃度を低下させるとcdna合成における異常な内部プライミングをある程度抑制することができるが,これを完全に排除することは困難であることが分かっている。 したがって、全体的なプロトコルの実証のために二ラウンドのcRNA増幅を行ったが、実際には、第一ラウンドの反応で十分なcRNAが得られる場合には、第二ラウンドのcrna増幅をスキップすることをお勧めします。
これらの結果を考慮すると、我々は我々の新しい方法は、少量の出発RNAからcDNAライブラリ構築に有用であると結論します。 実際、本発明者らは、全RNAの量が1μ g未満である場合に、cDNAライブラリーの構築のためにこの方法を正常かつ日常的に使用した(データは示されていない)。 1μ gの総RNAは、105-106哺乳動物細胞または1mgの哺乳動物組織から回収することができるので、我々の方法を用いて細胞選別機またはマイクロディセクション さらに、この方法は、単一細胞中の総RNAの量に近い105pg総RNAから1以上のcDNAクローンを得ることができることを計算するため、単一細胞由来cDNAライブラリー
謝辞
三沢和治博士からの統計解析の指導、古賀久博士からのプラスミドDnaの部門、浮貝明子氏と渡辺隆氏からの技術的支援に感謝しています。 この研究は、上総DNA研究所からR.O.、R.F.K.、O.O.
競合する利益声明
著者らは競合する利益を宣言していません。
- 1. Carninci,P.,C.Kvam,北村A.,大隅T.,岡崎Y.,伊藤M.,神谷M.,柴田K.et al.. 1996. ビオチン化キャップトラッパーによる高効率フルレングスcDNAクローニング。 ゲノミクス37:327-336。Crossref,Medline,CAS,Google Scholar
- 2. 小原大,長瀬大,石川大,中嶋大,大平大,関大,野村大,中嶋大,中嶋大,中嶋大,中嶋大,中嶋大,中嶋大,中嶋大,中嶋大,中嶋大,中嶋大 1997. 比較的大きなタンパク質をコードするcDNAクローンの分析に適したヒト脳cDNAライブラリーの構築と特性評価。 DNA Res.4:53-59。Crossref,Medline,CAS,Google Scholar
- 3. 鈴木,吉友-中川,丸山,須山,菅野,須賀野,須賀野,須賀野,須賀野,須賀野,須賀野,須賀野,須賀野,須賀野,須賀野,須賀野 1997. 完全長濃縮および5’末端濃縮cDNAライブラリーの構築および特性評価。 遺伝子200:1149-1156。Crossref,Google Scholar
- 4. 大原、O.およびG.寺院。 2001. Invitro組換え反応によって支援される方向性cdnaライブラリー構築。 Nucleic Acids Res.Crossref,Medline,CAS,Google Scholar
- 5. Eberwine,J.,H.Yeh,K.Miyashiro,Y.Cao,S.Nair,R.Finnel,M.Zettel,P.Coleman. 1992. 単一の生きているニューロンの遺伝子発現の分析。 プロク… ナトル アカド サイ… 米国89:3010-3014。Crossref,Medline,CAS,Google Scholar
- 6. Wang,E.,L.D.Miller,G.A.Ohnmacht,E.T.Liu,And F.M.Marincola. 2000. 遺伝子のプロファイリングのための高忠実度のmRNAの拡大。 ナット バイオテクノール 18:457–459.Crossref,Medline,CAS,Google Scholar
- 7. ボー、L.R.、A.A.ヒル、E.L.ブラウン、C.P.ハンター。 2001. In vitro転写によるmRNA増幅の定量分析。 Nucleic Acids Res.Crossref,Medline,CAS,Google Scholar
- 8. Jumabayeva、B.、L.Diatchenko、A.Chenchik、およびP.D.Siebert。 2001. 複数のヒト腫瘍における遺伝子発現研究のためのSMART™生成cDNAの使用。 BioTechniques30:158-163.リンク、CAS、Google Scholar
- 9。 Gomes,L.,R.Silva,B.S.Stolf,E.B.Cristo,R.Hirata,Jr.,F.A.Soares,L.Reis,E.J.Neves,et al.. 2003. CDNAマイクロアレイにおけるハイブリダイゼーションのための増幅および非増幅RNAの比較分析。 アナル… バイオケム 321:244–251.Crossref、Medline、CAS、Google Scholar
- 10。 およびS. 2003. 切断されたBcl-2、前立腺癌の潜在的な前metasticマーカー。 バイオケム バイオフィス・バイフィス・バイフィス コミュ… 306:912–917.Crossref、Medline、CAS、Google Scholar
- 11。 Lin、S.L.およびS.Y.Ying。 2003. RNA-polymeraseサイクリング反応を用いたmRNA/cDNAライブラリー構築。 メソッドMol。 バイオル 221:129–143.Medline、CAS、Google Scholar
- 12。 マッツ、M.V.2003。 顕微鏡的な量の動物組織からの代表的なcDNAプールの増幅。 メソッドMol。 バイオル 221:103–116.Medline、CAS、Google Scholar
- 13。 Petalidis,L.,S.Bhattacharyya,G.A.Morris,V.P.Collins,T.C.Freeman,p.A.Lyons. 2003. テンプレートスイッチングPCRによるmRNAのグローバル増幅:線形性とマイクロアレイ解析への応用。 Nucleic Acids Res.Crossref,Google Scholar
- 14. Polacek,D.C.,A.G.Passerini,C.Shi,N.M.Francesco,E.Manduchi,G.R.Grant,S.Powell,H.Bischof,et al.. 2003. 内皮mRNAのナノグラム量の線形増幅後の差動転写プロファイルの忠実度と強化された感度。 フィジオール ゲノミクス13:147-156。Crossref、Medline、CAS、Google Scholar
- 15。 ウィルソン,C.S.D.Pepper,Y.こんにちは、C.J.メンバーをサポートしています。 2004. 増幅プロトコルは、遺伝子発現研究に系統的ではあるが再現性のあるエラーを導入する。 BioTechniques36:498-506.リンク、CAS、Google Scholar
- 16。 小原大-長瀬智也-G. 三井,幸賀博,菊野理,平岡理,高橋由,北島理,他. 2002. In vitro組換え支援法によって生成されたサイズ分画cDNAライブラリーのキャラクタリゼーション。 DNA Res.9:47-57。Crossref、Medline、CAS、Google Scholar
- 17。 大原大2003. サイズ分画したcdnaライブラリーの構築はinvitro組換え反応により支援した。 メソッドMol。 バイオル 221:59–71.Medline、CAS、Google Scholar
- 18。 Gubler、U.およびB.J.Hoffman。 1983. CDNAライブラリーを生成するための簡単で非常に効率的な方法。 遺伝子25:263-269。Crossref、Medline、CAS、Google Scholar
- 19。 Sambrook,J.and D.W.Russell. 2001. SDSによるアルカリ溶解によるプラスミドDNAの調製、p.1.31–1.42。 分子クローニング、(第3版)。 CSH Laboratory Press,Cold Spring Harbor,NY.Google Scholar
- 20。 Church,G.M.and W.Gilbert. 1984. ゲノムシークエンシング。 プロク… ナトル アカド サイ… 米国81:1991-1995。Crossref、Medline、CAS、Google Scholar
- 21。 とができることを示しています。 2000. 変異ポリアデニル化のパターンは、ヒト遺伝子における使用を示している。 ゲノムRes.10:1001-1010.Crossref、Medline、CAS、Google Scholar
- 22。 タナカ,T.S.,S.A.Jaradat,M.K.Lim,G.J.Kargul,X.Wang,M.J.Grahovac,S.Pantano,Y.Sano,et al.. 2000. 15,000マウス発達cDNAマイクロアレイを使用して妊娠中期胎盤と胚のゲノムワイド発現プロファイリング。 プロク… ナトル アカド サイ… 米国97:9127-9132。Crossref、Medline、Google Scholar
- 23。 ルキヤノフ,K.,L.Diatchenko,A.Chenchik,A.Nanisetti,P.Siebert,N.Usman,M.Matz,S.Lukyanov. 1997. 抑制PCR効果を用いた少量の全RNAからのcDNAライブラリーの構築。 バイオケム バイオフィス・バイフィス・バイフィス コミュ… 230:285–288.Crossref、Medline、CAS、Google Scholar
- 24。 Piao,Y.,N.T.Ko,M.K.Lim,M.Ko. 2001. 普遍的なPCR増幅法による総Rnaのサブミクロン量からの長い転写物濃縮cDNAライブラリーの構築。 ゲノムRes.11:1553-1558.Crossref,Medline,CAS,Google Scholar