
Introduction
Le sequenze complete del genoma di vari organismi, compresi i mammiferi, sono diventate recentemente disponibili come conseguenza dei rapidi progressi nella tecnologia di sequenziamento del DNA. Tuttavia, in particolare nei mammiferi, l’analisi dei trascritti svolge ancora un ruolo chiave nel colmare il divario tra il genoma e il proteoma. Questo è principalmente perché, al momento, non possiamo prevedere con precisione le strutture dei trascritti derivati da un particolare gene dalle sole informazioni genomiche. Pertanto, come metodo per l’analisi delle trascrizioni, la costruzione della libreria cDNA è cruciale, anche nell’era del sequenziamento post-genoma. Anche se la clonazione cDNA di geni di interesse da PCR ha fornito un percorso alternativo semplificato per analizzare le strutture di trascrizione senza costruzione libreria cDNA, la costruzione di una libreria cDNA è l’approccio di scelta quando un gran numero di CDNA da una singola fonte di mRNA devono essere analizzati. Ad oggi, è stato fatto un grande sforzo nello sviluppo di un metodo per la preparazione di librerie cDNA di alta qualità (1-4). Al contrario, la questione di come preparare una libreria di cDNA di alta qualità da un piccolo pool di RNA non è stata affrontata in modo così attivo. Tuttavia, questo è diventato un obiettivo ad alta priorità perché i ricercatori sono spesso interessati a geni ipotetici che si prevede siano espressi solo in alcuni tipi di cellule o tessuti in condizioni particolari, come quelli osservati in campioni patologici.
Ci sono stati un certo numero di rapporti che descrivono l’uso di piccole quantità di RNA sorgente per generare cDNA amplificato o cRNA, da cui possono essere preparati obiettivi per l’analisi microarray (5-15). Il loro obiettivo finale è quello di ottenere RNA a lunghezza intera, non polarizzato dalla popolazione, altamente rappresentativo dell’mRNA originale. Per eseguire questo, questi metodi di solito adottano PCR o trascrizione in vitro da T7 RNA polimerasi per amplificare l’mRNA originale sotto forma di cDNA o cRNA, rispettivamente. Sebbene il confronto dei profili di espressione sia uno degli approcci più efficienti per indagare le differenze negli stati fisiologici delle cellule o dei tessuti, la caratterizzazione strutturale dei trascritti (ad esempio, identificazione di modelli di splicing alternativi, sito di inizio trascrizione e sito di terminazione della trascrizione) non può essere eseguita da tale analisi microarray. L’analisi di sequenziamento di ogni trascrizione è inevitabile in questi casi.
Abbiamo sviluppato un metodo che consente di utilizzare una piccola quantità di RNA iniziale per costruire una libreria di cDNA adatta alla clonazione genica e all’analisi completa del sequenziamento. Il metodo etichetta le estremità 3′ e 5′ dei CDNA del primo round con sequenze di promotori di fagi T7 e SP6, rispettivamente, per ridurre al minimo gli effetti di polarizzazione delle dimensioni durante l’amplificazione. Dopo l’amplificazione cRNA a due giri, abbiamo convertito i CRNA amplificati in CDNA, clonato questi prodotti in un plasmide e quindi valutato la libreria cDNA risultante rispetto a quella costruita con un metodo convenzionale (4,16,17).
Materiali e metodi
Amplificazione del cRNA
Le sequenze di oligonucleotidi utilizzate in questo studio sono riportate nella Tabella 1. Per stabilire il protocollo, abbiamo usato 1 µg di RNA totale preparato dal cervello di topo ICR (topi maschi di 8 settimane) come modello. Una miscela(10 µL) di 1 µg di RNA totale e 100 pmol T7-Not-(DT)18 primer è stata incubata a 70°C per 10 min e poi raffreddata a scatto su ghiaccio. La sintesi di cDNA a doppio filamento e la legatura dell’adattatore sono state eseguite seguendo i protocolli del sistema plasmidico SUPERSCRIPT® (Invitrogen, Carlsbad, CA, USA) (4,16,17), con piccole modifiche. In breve, per la sintesi del cDNA di primo filamento, 4 µL 5× tampone di primo filamento (Invitrogen), 1 µL 0,1 M ditiotreitolo (DTT), 1 µL 10 mm dNTPs, 1 µL (40 U) RNaseOUT™ (Invitrogen) e 1 µL di acqua sono stati aggiunti alla miscela di primer RNA/T7-Not-(dT)18 denaturato e incubati a 37°C per 3 min per Quindi, 2 µL (400 U) APICE III RNasi H-trascrittasi inversa (Invitrogen) sono stati aggiunti alla miscela di reazione e la temperatura è stata regolata a 50°C per avviare la sintesi del cDNA di primo filamento. Dopo 1 h, il secondo-strand cDNA di sintesi è stata effettuata come descritto in precedenza da Gubler e Hoffman (18); per il first-strand cDNA miscela, 91 µL di acqua, 30 µL 5× seconda-strand buffer (Invitrogen), 3 µL 10 mM dNTPs, 1 µL (10 U) Escherichia coli DNA ligasi, 4 µL (40 U) E. coli DNA polimerasi, e 1 µL (2 U) E. coli Rnasi H sono stati aggiunti, e la miscela è stata incubata a temperatura di 16°C per 2 h. cDNA termini erano quindi fine lucidato con 10 U T4 DNA polimerasi (Invitrogen) a 16°C per 5 min, e la reazione è stata interrotta con l’aggiunta di 10 µL 0,5 M etilendiammina acido etilendiamminotetraacetico (EDTA). I CDNA risultanti sono stati purificati mediante estrazione con fenolo/cloroformio / alcool isoamilico (25:24:1), seguita da precipitazione di etanolo come descritto in precedenza (17), ad eccezione dell’uso di 1 µg di lievito tRNA invece di acido poliadenilico come vettore. Il cDNA precipitato è stato sciolto in 50 µL di TE (10 mM Tris-HCl, pH 8,0 e 1 mM EDTA), mescolato con 30 µL di una soluzione di NaCl al 20% (p/v) di polietilenglicole (PEG) 6000/2, 5 M e quindi incubato su ghiaccio per 1 h (17). Dopo l’incubazione, la miscela di cDNA è stata centrifugata a 18.000× g a 4°C per 15 min. Il pellet di cDNA risultante è stato risciacquato due volte con etanolo al 70%, essiccato e quindi risospeso in acqua 30 µL. I CDNA purificati sono stati ligati con un adattatore SP6 da 500 pmol (Tabella 1) utilizzando 5 U T4 DNA ligasi (Invitrogen) in un volume di reazione di 50 µL a 16°C durante la notte. La miscela legata all’adattatore è stata diluita due volte con acqua, quindi i CDNA sono stati purificati utilizzando una colonna DNAclear™ (Ambion, Austin, TX, USA). L’eluato (in 16 µL di acqua) è stato utilizzato come modello per la sintesi di cRNA. Utilizzando il kit MEGAscript® T7 (Ambion), i CRNA sono stati sintetizzati a 37°C durante la notte in un volume di reazione di 40 µL. Dopo che i CDNA modello sono stati degradati da 4 U DNasi I, i CRNA sintetizzati sono stati purificati utilizzando un mini kit RNeasy® (QIAGEN, Valencia, CA, USA) ed eluiti con acqua 100 µL. La concentrazione del cRNA è stata determinata dall’assorbimento ultravioletto (UV). Per l’amplificazione del cRNA secondo round, 2 µg del CRNA sintetizzato e 100 pmol SP6 UP primer (pari all’oligonucleotide del filamento superiore dell’adattatore SP6 mostrato nella Tabella 1) sono stati utilizzati rispettivamente come template e primer. La sintesi di cDNA secondo turno è stata effettuata come nella trascrizione inversa originale dall’RNA sorgente, tranne che SP6 UP primer ricottura è stata eseguita a 50°C per 3 min. Dopo la purificazione del cDNA a doppio filamento ottenuto attraverso una colonna DNAclear, 0,5 µg del cDNA sono stati utilizzati come modello per la successiva amplificazione CRNA assistita da SP6 RNA polimerasi. Utilizzando il kit MEGAscript SP6 (Ambion), la sintesi del cRNA è stata effettuata a 37°C per 6 h. Dopo che il modello cDNA è stato degradato usando DNasi I, i CRNA sintetizzati sono stati purificati come descritto sopra.
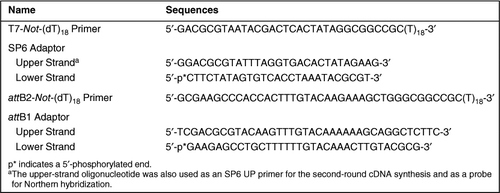
Costruzione di librerie cDNA
Due microgrammi dei CRNA risultanti sono stati sottoposti a sintesi di cDNA a doppio filamento utilizzando 100 pmol attB2-Not-(dT)18 primer (Tabella 1). La sintesi del cDNA, l’otturazione finale, la purificazione e le fasi di legatura dell’adattatore sono state eseguite come nelle precedenti sintesi del cDNA, tranne per il fatto che l’adattatore attB1 (Tabella 1, 500 pmol) è stato legato ai CDNA a doppio filamento. Dopo che i CDNA ATTB1 legati all’adattatore sono stati purificati da trattamenti successivi di estrazione di fenolo/cloroformio / alcool isoamilico, precipitazione di etanolo e precipitazione PEG/NaCl in questo ordine, sono stati sciolti in 50 µL TE e trattati con RNasi A (ad una concentrazione finale di 10 µg/mL; Invitrogen) per degradare l’RNA contaminato a 37°C per 30 min. La miscela di reazione è stata nuovamente purificata mediante estrazione con fenolo/cloroformio / alcool isoamilico, seguita da precipitazione di etanolo e precipitazione di PEG/NaCl. I CDNA risultanti sono stati sciolti in 15 µL di TE e la loro quantità è stata stimata dall’intensità della colorazione fluorescente dopo elettroforesi su gel di agarosio (4). I CDNA attB1-ligati (36 ng) sono stati sottoposti ad una reazione di ricombinazione in vitro (Gateway® System; Invitrogen) con 250 ng attP-pSP73 vettore donatore come descritto in precedenza (4,16,17). In breve, il vettore donatore attB1-legated cDNA e attP-pSP73 sono stati miscelati e incubati in un volume di reazione di 10 µL contenente 2 µL 5× tampone di reazione BP Clonase™ e 2 µL BP Clonase (entrambi da Invitrogen) a 25°C durante la notte. La miscela di cDNA è stata trattata con 2 µg di proteinasi K (Invitrogen) a 37°C per 10 min per estinguere la reazione ed estratta con fenolo/cloroformio/alcool isoamilico, seguita da precipitazione di etanolo con 2 µg di lievito tRNA come vettore. Il cDNA precipitato è stato sciolto in 10 µL di acqua e 1 µL della miscela è stata utilizzata per la trasformazione delle cellule di E. coli ElectroMAX™ DH10B™ (Invitrogen). Dopo la titolazione della libreria cDNA risultante, i plasmidi cDNA sono stati recuperati da circa 1.000.000 di colonie con un metodo alcalino di sodio dodecil solfato (SDS) come descritto in precedenza (4,19). I cloni cDNA risultanti erano in una forma di plasmidi che trasportavano siti attL. Poiché i plasmidi che trasportano siti attB sono necessari in alcune applicazioni, questi plasmidi sono stati convertiti in quelli che trasportano siti attB come precedentemente descritto (16). Questa libreria cDNA è stata di conseguenza designata come MB-AL (mouse brain amplified CRNA-derived library).
Nello stesso modo descritto per MB-AL, abbiamo costruito una libreria cDNA da RNA nonamplificato (100 µg di RNA totale del cervello del topo) e l’abbiamo designata come MB-CL (mouse brain convenzionalmente costruito biblioteca).
Esame di Dimensione-Bias Effetto Amplificato cRNAs
esaminare la dimensione bias effetto cRNAs, il primo turno delle cRNAs trascritto da T7 RNA polimerasi e il secondo turno cRNAs trascritto da SP6 RNA polimerasi sono stati eseguiti su 1.0% gel di agarosio (1 µg ciascuna) e poi cancellati su membrana di nylon (Biodyne® B Membrana di Nylon; PALL, East Hills, NY, USA). Sono stati ibridati con 32P-marcato SP6 UP primer (Tabella 1)e Non-(dT) 18 (5′-GCGGCCGCTTTTTTTTTTTTTTTTTTTT-3′) oligonucleotide, rispettivamente. Dieci picomoli di ciascuna sonda sono stati marcati con ATP (circa 3000 Ci / mmol; Amersham Biosciences, Piscataway, NJ, USA) utilizzando la polinucleotide chinasi T4 (Takara, Kyoto Giappone). Dopo ibridazione notturna in tampone GMC (20) a 45°C, queste membrane sono state ampiamente lavate in soluzione salina 1× standard citrato (SSC)/1% SDS tre volte a temperatura ambiente. L’analisi dei segnali ibridati è stata eseguita su un analizzatore di immagini BAS 2000 (Fuji Photo Film, Tokyo, Giappone)
Valutazione delle librerie cDNA
Elettroforesi su gel. La distribuzione delle dimensioni dell’inserto del cDNA è stata esaminata mediante elettroforesi di CRNA sintetizzati dalla T7 RNA polimerasi utilizzando CDNA plasmidici MB-AL e MB-CL non digeriti come modelli. Dopo la purificazione con un mini kit RNeasy, i CRNA sono stati elettroforesi su un gel di agarosio contenente formaldeide con marcatori di RNA perfetti (EMD Biosciences, Madison, WI, USA). Dopo la colorazione del gel con SYBR ® Green II( Invitrogen), l’intensità della colorazione fluorescente è stata analizzata utilizzando il software ImageQuant® (versione 5.0) su un FluorImager 595 (Amersham Biosciences).
Analisi di sequenziamento casuale a passaggio singolo. 3 ‘ – End sequenze di cloni cDNA sono stati esaminati mediante sequenziamento del DNA. Il DNA plasmidico di 768 cloni scelti casualmente è stato purificato utilizzando un Magnia ® MFX-9600 (Toyobo, Osaka, Giappone) e analizzato da un sequencer RISA 384-Capillare (Shimadzu, Kyoto, Giappone) (16). Dopo aver tagliato la sequenza vettoriale utilizzando il software Sequencher™ versione 4.1 (Software Hitachi, Tokyo, Giappone), le sequenze di cDNA ottenute che erano più lunghe di 200 residui di nucleotidi sono state raggruppate con voci di cDNA nel database GenBank® e nel nostro database di mouse interno. L’esame dell’integrità delle estremità 3′ dei cloni di cDNA è stato eseguito analizzando se un esamero canonico del segnale di poliadenilazione (5′-AATAAA-3’) o le sue varianti a base singola (11 esameri del segnale) sono stati trovati all’interno dei residui di 50 nucleotidi a monte della coda di poli(A) dei cDNA (21).
cDNA microarray. Sono state eseguite analisi di microarray per confrontare le popolazioni di cDNA nelle librerie MB-AL e MB-CL. Sono stati preparati microarray in nylon cDNA fatti in casa con sonde 3534 ed è stato eseguito il rilevamento radioisotopico (22). In breve, i plasmidi cDNA, che sono stati isolati nel nostro istituto e per i quali erano già note sequenze complete o sequenze finali, sono stati individuati utilizzando GeneTAC ™ RA1 (Genomic Solutions, Ann Arbor, MI, USA) su membrana di nylon Biodyne B e quindi fissati seguendo le istruzioni del produttore. L’elenco dei CDNA immobilizzati su questo microarray è disponibile su richiesta degli autori. I CDNA target MB-AL e MB-CL sono stati preparati utilizzando la trascrizione inversa da ciascuno dei campioni di cRNA da 1 µg utilizzati per l’analisi delle dimensioni dell’inserto come descritto sopra. Questi obiettivi sono stati sintetizzati ed etichettato con una miscela di reazione contenente 1 µg di ogni cRNA, 100 ng casuale hexamer, 1× first-strand buffer, 10 mM DTT, ogni 800 µM dATP, dTTP, e dGTP, 800 nM dCTP, 5 µL di dCTP (>2500 Ci/mmol; Amersham Biosciences), e 100 U SuperScript II Rnasi H – trascrittasi inversa a temperatura ambiente per 10 min e poi a 42°C per 1 h. Dopo la lisi alcalina e di neutralizzazione, l’etichetta cdna sono stati purificati con il QIAquick® kit di purificazione PCR (QIAGEN) e contati. Il cDNA target è stato sottoposto ad ibridazione con il cDNA nylon microarray in presenza di 1 µg di acido poliadenilico e 2,5 µg di Mouse Cot-1 DNA® (Invitrogen) in 250 µL di PerfectHyb™ buffer (Toyobo) a 68°C durante la notte. Dopo un lavaggio rigoroso a 68°C con 2× SSC/1% SDS (due lavaggi da 15 min ciascuno) e poi con 0,1× SSC/1% SDS (due lavaggi da 30 min ciascuno), i segnali di ibridazione sono stati rilevati e analizzati su un FLA-8000 (Fuji Photo Film). I punti cDNA che mostrano segnali più forti di quelli del controllo di fondo sono stati selezionati e sottoposti a ulteriori analisi. Dopo aver effettuato la normalizzazione globale, sono stati ottenuti grafici a dispersione delle intensità del segnale dei punti cDNA provenienti dagli obiettivi MB-AL e MB-CL.
Ibridazione della macchia di RNA. RNA blot ibridazione è stata effettuata per esaminare gli effetti dimensione-bias. I CRNA MB-AL e MB-CL (ciascuno 1,5 µg, sintetizzati come descritto sopra)sono stati elettroforesi su gel di agarosio contenente formaldeide e trasferiti alla membrana di nylon Biodyne B. I CDNA della sonda sono stati preparati utilizzando l’amplificazione PCR. I primer sono stati progettati in base alle sequenze registrate nel database GenBank o nel nostro database interno; i modelli erano i plasmidi cDNA che avevamo isolato. La sonda della proteina ribosomiale S6 era lunga 724 bp, amplificata con primer 5′-CGCTCGGCTGTGTCAAGATG-3’e 5′-GAGGACAGCCTACGTCTCTTGG-3 ‘e la sonda della proteina da shock termico (hsp) lunga 70.940 bp, è stata amplificata con 5′-GATGGACAAGGCGCAGATCC-3′ e 5’-CTCGATGGGGGGTCCTGAGC-3 ‘ primer. Queste sonde sono state etichettate con dCTP (circa 3000 Ci/mmol; Amersham Biosciences) utilizzando il sistema di etichettatura del DNA RadPrime (Invitrogen). Dopo ibridazione notturna in tampone PerfectHyb a 65°C, le membrane sono state lavate successivamente con 0,1× SSC/1% SDS a temperatura ambiente per 5 min e per 15 min, e poi a 65°C per 30 min. I segnali di ibridazione sono stati rilevati su un analizzatore di immagini BAS 2000.
Risultati e discussione
Il metodo di costruzione della libreria cDNA che abbiamo sviluppato in questo studio è mostrato in Figura 1 (i passaggi appena introdotti sono evidenziati con una stella). Questo metodo consiste di due cicli di fase di amplificazione cRNA, conversione di CRNA in CDNA, e la clonazione ricombinazionale in un plasmide. Il passo chiave in questo metodo è la legatura adattatore per tag cDNA termina con sequenza promotore SP6. Una volta che possiamo sintetizzare con successo i CDNA in questo formato, diventa possibile trascrivere in modo specifico i CRNA del primo round che contengono la sequenza del promotore SP6 alla loro estremità 3′. I CRNA di secondo turno sono sintetizzati con SP6 RNA polimerasi utilizzando i CDNA a doppio filamento risultanti come modelli. Dopo i due cicli di amplificazione assistita da RNA polimerasi, i CRNA risultanti vengono quindi convertiti in CDNA a doppio filamento mediante trascrizione inversa utilizzando il primer attB2-Not-(dT)18. L’uso del primer attB2-Not-(dT)18 nella trascrizione inversa ci consente di convertire solo i CRNA che trasportano la sequenza 5′-(A)18GCGGCCGC-3′ alle loro estremità 3′. Poiché queste modifiche dovrebbero consentire solo l’amplificazione e la clonazione di CDNA che hanno estremità correttamente etichettate, ci aspettiamo che questo metodo minimizzi il bias delle dimensioni durante l’amplificazione assistita da RNA polimerasi.

Sono illustrati i passaggi dell’amplificazione CRNA assistita da adattatore-legatura e la seguente costruzione della libreria cDNA. Per mantenere le dimensioni dei cDNA amplificati con quelle dei CDNA di primo turno, abbiamo introdotto alcune modifiche nei metodi convenzionali assistiti da RNA polimerasi (evidenziati con stelle). Le linee blu e rosse indicano rispettivamente cDNA e RNA. E. coli, Escherichia coli.
Per testare l’efficacia del metodo, abbiamo costruito librerie cDNA con un metodo convenzionale (16) utilizzando RNA totale nonamplificato dal cervello di topo (100 µg) e con il metodo sopra descritto utilizzando 1 µg di RNA totale dal cervello di topo. La tabella 2 riassume le quantità di cDNA e cRNA ottenute in ogni fase, calcolate come una media di tre cicli sperimentali indipendenti di costruzione MB-AL. Abbiamo ottenuto una media di 6,4×107 cloni cDNA indipendenti come output finale. Poiché questo numero di cloni di cDNA deriva da un totale di 36 ng cDNA generati utilizzando il protocollo di amplificazione, calcoliamo che questo metodo produce circa 1,2×1011 cloni di cDNA da 1 µg di RNA totale.

Per esaminare sperimentalmente l’effetto di polarizzazione delle dimensioni durante le fasi di amplificazione, abbiamo successivamente confrontato le distribuzioni delle dimensioni del cRNA di primo e secondo turno mediante analisi di ibridazione RNA blot. In questi esperimenti, i CRNA di primo e secondo turno sono stati ibridati con primer SP6 UP e con oligonucleotide Not-(dT)18, rispettivamente, perché queste sonde oligonucleotidiche rilevano solo CRNA correttamente trascritti fino alla fine. Come mostrato in Figura 2, la dimensione dell’RNA al picco del segnale di ibridazione nel cRNA di secondo turno è risultata essere quasi la stessa di quella del cRNA di primo turno, ma con un leggero spostamento verso dimensioni più piccole. Sospettiamo che il troncamento dei CRNA abbia probabilmente avuto luogo durante la sintesi del cDNA di secondo turno con il primer SP6 UP.

RNA blot ibridazione è stata effettuata per esaminare dimensione-bias effetto sui CRNA. Sono stati tracciati segnali di ibridazione lungo la direzione dell’elettroforesi del cRNA. La dimensione dell’RNA è stata determinata in base alla mobilità del marcatore della scala dell’RNA. Linea blu, il cRNA primo turno ibridato con SP6 UP sonda; linea rossa, il cRNA secondo turno ibridato con Not-(dT)18 sonda; valore PSL, intensità del segnale indicato arbitrariamente dal Software Calibro immagine (Fuji Photo Film).
Per valutare ulteriormente la qualità della libreria cDNA amplificata risultante, abbiamo anche confrontato varie caratteristiche delle librerie cDNA generate con e senza l’amplificazione (MB-AL e MB-CL, rispettivamente). Abbiamo prima esaminato la distribuzione dimensionale degli inserti cDNA, come mostrato in Figura 3A. I risultati dei segnali di intensità della fluorescenza ottenuti dall’immagine di colorazione del gel hanno indicato che le dimensioni degli inserti cDNA di MB-AL e MB-CL erano simili, ma il punto di picco era leggermente più piccolo in MB-AL (0,65 kb) rispetto a quello in MB-CL (0,8 kb). Successivamente, abbiamo analizzato 3 ‘ – end sequenze di cloni cDNA campionati in modo casuale da ogni libreria. I risultati del clustering per questi expressed Sequence tag (EST) sono mostrati in Figura 3B e suggeriscono che la complessità di MB-AL era alta quanto quella di MB-CL. Tuttavia, vale la pena notare che i cloni cDNA altamente ridondanti sono scomparsi in MB-AL. Da un’analisi statistica (test Chi), è stato dimostrato che la differenza di ridondanze tra MB-AL e MB-CL era significativa (la probabilità che le ridondanze in MB-AL e MB-CL siano simili è <0.001). Inoltre, abbiamo esaminato l’integrità delle estremità 3’ dei CDNA analizzando se ogni EST contenesse un esamero del segnale di poliadenilazione o meno. I risultati hanno indicato che l ‘80,1% e l’ 87,3% dei cloni di cDNA in MB-AL e MB-CL, rispettivamente, contenevano sequenze di segnali di poliadenilazione plausibili (21). La diminuzione del tasso di occorrenza degli esameri del segnale di poliadenilazione in MB-AL ha probabilmente indicato un aumento del numero di cloni di cDNA in cui la sintesi di cDNA è stata innescata internamente a causa di due cicli di adescamento dT.

MB-AL (mouse brain amplified CRNA-derived library) è stato valutato in confronto con una libreria costruita convenzionalmente, MB-CL. (A) Confronto delle lunghezze dei cDNA degli inserti. I CRNA trascritti in vitro sono stati frazionati in dimensioni su gel di agarosio e macchiati. Le loro intensità fluorescenti lungo la direzione dell’elettroforesi (indicata come dimensione dell’RNA) sono mostrate in unità fluorescenti relative (RFU). (B) Risultati di clustering di MB-AL e MB-CL 3′-end sequenze (EST). (C) Confronto della popolazione di cDNA in MB-AL e MB-CL. Sono stati tracciati segnali ottenuti mediante ibridazione microarray. (D) Risultati di ibridazione di RNA blot sulla proteina ribosomiale S6 e hsp 70. I CRNA MB – AL erano nella corsia di sinistra e quelli di MB-CL erano nella corsia di destra (indicati rispettivamente da AL e CL). Le punte di freccia indicano le posizioni della dimensione prevista dell’RNA di ciascun gene. hsp, proteina da shock termico.
Mediante ibridazione microarray, abbiamo esaminato la popolazione cDNA di MB-AL rispetto a quella di MB-CL. La figura 3C mostra un grafico a dispersione dei segnali di ibridazione degli obiettivi MB-AL e MB-CL. I risultati hanno indicato una correlazione considerevolmente elevata dei segnali di ibridazione tra obiettivi MB-AL e MB-CL, suggerendo che non vi era un effetto dannoso dell’amplificazione nella popolazione di cDNA.
Infine, abbiamo eseguito l’analisi di ibridazione RNA blot per esaminare gli effetti di dimensione-bias con attenzione ai geni di housekeeping (Figura 3D). Per la proteina ribosomiale S6 e l’hsp 70, le dimensioni del cRNA sia in MB-AL che in MB-CL erano simili e comparabili a quelle attese dalle sequenze nucleotidiche riportate. Tuttavia, il modello di ibridazione dei CRNA hsp 70 in MB-AL era leggermente diverso da quello in MB-CL, indicando che l’effetto di polarizzazione delle dimensioni non era completamente rimosso.
Il metodo di costruzione della libreria cDNA descritto in questo rapporto è stato ideato per ridurre al minimo i pregiudizi di dimensione durante le fasi di amplificazione assistite dalla RNA polimerasi. Tale metodo di costruzione della biblioteca assistito da RNA polimerasi da un piccolo pool di RNA per un’analisi di sequenziamento completa non è stato ben studiato, mentre i metodi di costruzione della biblioteca cDNA/cRNA per l’ibridazione microarray sono stati attivamente perseguiti. Anche se Lukyanov et al. (23) e Piao et al. (24) metodi riportati utilizzando l’amplificazione PCR per costruire una libreria di cDNA a partire da una quantità submicrogramma di RNA totale, l’amplificazione PCR presenta intrinsecamente degli svantaggi, come una grave distorsione delle dimensioni e della popolazione e una bassa fedeltà dell’amplificazione cDNA. Questo è stato il motivo per cui abbiamo cercato di sviluppare un metodo di costruzione della libreria cDNA basato sull’amplificazione assistita da RNA polimerasi in questo studio.
A questo scopo, abbiamo ideato una nuova strategia di amplificazione assistita da RNA polimerasi come mostrato in Figura 1 perché i metodi di Eberwine et al. (5), Huang et al. (10), e Lin et al. (11) ancora possiedono gli svantaggi per la preparazione di un cDNA plasmid-contenuto adatto ad analisi completa della struttura del gene. Nel primo, la sintesi di cDNA dopo l’amplificazione del cRNA viene eseguita con esameri casuali, e questo rende i CDNA amplificati inevitabilmente più brevi di quelli originali (5,7). In quest’ultimo, l’introduzione di una coda omopolimerica all’estremità del cDNA del primo filamento da parte della deossinucleotidil transferasi terminale (TdT) o del virus della leucemia murina di Moloney (MMLV) attività inversa della trascrittasi (10,11) ha senso ridurre al minimo l’effetto di polarizzazione delle dimensioni durante l’amplificazione assistita da RNA polimerasi come nel nostro metodo. Tuttavia, il metodo di tailing omopolimerico è noto per causare il troncamento dei CDNA a causa dell’innesco inaspettato della sintesi di cDNA da un tratto omopolimerico di sequenze di RNA interne. Poiché il metodo di legatura dell’adattatore descritto in questo studio potrebbe fornire una sequenza specifica per l’adescamento del cDNA, potremmo aspettarci di ridurre l’entità del troncamento del cDNA usando il nostro metodo.
In linea di principio, il nostro metodo dovrebbe solo amplificare i CDNA affiancati da una coda intrinseca di poli(A) e dalla sequenza di adattatori SP6, che sono stati generati nella sintesi del cDNA di primo turno. I risultati mostrati nella figura 2 e nella figura 3A erano sostanzialmente coerenti con questo, sebbene la popolazione di cDNA nella gamma di dimensioni più piccole fosse leggermente aumentata. I risultati dell’analisi RNA blot (Figura 3D) hanno anche mostrato che l’effetto dimensione-bias potrebbe essere ridotto ma non completamente rimosso anche nel nostro metodo. Ciò è stato probabilmente dovuto al fatto che i CDNA troncati aggiunti da una coda poli(A) e la sequenza del promotore SP6 sono stati generati durante la conversione di cRNA in cDNA. Questa visione era supportata dal fatto che il tasso di occorrenza delle sequenze di segnali di poliadenilazione in MB-AL era significativamente inferiore a quello osservato nella libreria cDNA convenzionale (21). Il troncamento dei CDNA potrebbe verificarsi durante la sintesi dei cDNA a causa dell’innesco interno dell’RNA con il primer DT-tailed e il primer SP6. Poiché gli stiramenti omopolimerici si verificano frequentemente negli MRNA eucariotici, il metodo di tailing omopolimerico potrebbe imporre un pregiudizio di dimensioni più gravi sull’amplificazione cRNA a più giri rispetto al nostro metodo di legatura dell’adattatore. Sebbene aumentare la temperatura di reazione durante la trascrizione inversa e diminuire la concentrazione di primer potrebbe sopprimere l’innesco interno aberrante nella sintesi di cDNA in una certa misura, è noto che è difficile eliminarlo completamente al momento. Così, anche se abbiamo effettuato due round di amplificazione cRNA per la dimostrazione del protocollo generale, in pratica, si consiglia di saltare il secondo round di amplificazione cRNA quando abbastanza cRNA può essere ottenuto nella reazione di primo round.
Considerando questi risultati, concludiamo che il nostro nuovo metodo è utile per la costruzione della libreria cDNA da una piccola quantità di RNA iniziale. Infatti, abbiamo usato con successo e di routine questo metodo per la costruzione di librerie cDNA quando la quantità di RNA totale è inferiore a 1 µg (dati non mostrati). Poiché 1 µg di RNA totale potrebbe essere recuperato da 105-106 cellule di mammifero o 1 mg di tessuti di mammifero, le librerie di cDNA possono essere facilmente costruite da cellule frazionate da selezionatori cellulari o microdissezione utilizzando il nostro metodo. Inoltre, poiché calcoliamo che questo metodo ci consente di ottenere più di 105 cloni di cDNA da 1 pg di RNA totale, che è vicino alla quantità di RNA totale in una singola cellula, una libreria di cDNA derivata da una singola cellula potrebbe essere costruita sulla base di questa amplificazione CRNA assistita da adattatore-legatura.
Ringraziamenti
Gli autori sono grati per l’istruzione di analisi statistica dal Dr. Kazuharu Misawa; divisione dei DNA plasmidici dal Dr. Hisashi Koga; e aiuto tecnico da Ms. Akiko Ukigai-Ando e Mr. Takashi Watanabe. Lo studio è stato supportato da una sovvenzione del Kazusa DNA Research Institute a R. O., R. F. K. e O. O.
Dichiarazione di interessi concorrenti
Gli autori non dichiarano interessi concorrenti.
- 1. Carninci, P., C. Kvam, A. Kitamura, T. Ohsumi, Y. Okazaki, M. Itoh, M. Kamiya, K. Shibata, et al.. 1996. Clonazione di cDNA full-length ad alta efficienza con trapper a CAPPUCCIO biotinilato. Genomica 37: 327-336.Crossref, Medline, CAS, Google Scholar
- 2. Ohara, O. T. Nagase, K. Ishikawa, D. Nakajima, M. Ohira, N. Seki, e N. Nomura. 1997. Costruzione e caratterizzazione di librerie di cDNA del cervello umano adatte all’analisi di cloni di cDNA che codificano proteine relativamente grandi. DNA Res. 4: 53-59.Crossref, Medline, CAS, Google Scholar
- 3. Il suo nome deriva dal latino “Yoshitomo-Nakagawa”, “K. Maruyama”, “A. Suyama”, “S. Sugano”, “K. Yoshitomo-Nakagawa”, “K. Maruyama”, “A. Suyama”. 1997. Costruzione e caratterizzazione di una libreria cDNA completa e arricchita da 5′. Gene 200: 1149-1156.Crossref, Google Scholar
- 4. Ohara, O. e G. Temple. 2001. Costruzione della libreria cDNA direzionale assistita dalla reazione di ricombinazione in vitro. Acidi nucleici Res. 29:e22.Crossref, Medline, CAS, Google Scholar
- 5. Eberwine, J. H., Yeh, K. Miyashiro, A. Cao, S. Nair, R. Finnel, M. Zettel, e P. Coleman. 1992. Analisi dell’espressione genica in singoli neuroni vivi. Proc. Natl. Acad. Sic. Stati Uniti D’America 89: 3010-3014.Crossref, Medline, CAS, Google Scholar
- 6. Il suo nome deriva dal greco antico, che significa “terra”, “terra”, “terra”, “terra”, “terra”, “terra”, “terra”, “terra”, “terra”, “terra”, “terra”, “terra”, “terra”, “terra”, “terra”. 2000. Ad alta fedeltà mRNA amplificazione per gene profiling. NAT. Biotecnologia. 18:457–459.Crossref, Medline, CAS, Google Scholar
- 7. Baugh, L. R., A. A. Hill, E. L. Brown, e C.P. Hunter. 2001. Analisi quantitativa dell’amplificazione dell’mRNA mediante trascrizione in vitro. Acidi nucleici Res. 29:e29.Crossref, Medline, CAS, Google Scholar
- 8. Zhumabayeva, B., L. Diatchenko, A. Chenchik e P. D. Siebert. 2001. Utilizzo di cDNA generati da SMART™per studi di espressione genica in più tumori umani. BioTechniques 30: 158-163.Link, CAS, Google Scholar
- 9. Il suo nome deriva dal greco antico, che significa “”Cristo””, “”Cristo””.. 2003. Analisi comparativa di RNA amplificato e non amplificato per ibridazione in cDNA microarray. Anale. Biochimica. 321:244–251.Crossref, Medline, CAS, Google Scholar
- 10. Il sito utilizza cookie tecnici e di terze parti. 2003. Bcl-2 troncato, un potenziale marcatore pre-metastico nel cancro alla prostata. Biochimica. Biophys. Res. Commun. 306:912–917.Crossref, Medline, CAS, Google Scholar
- 11. Lin, S. L. e S. Y. Ying. 2003. Costruzione della libreria mRNA / cDNA utilizzando la reazione di riciclaggio della RNA-polimerasi. Metodi Mol. Biol. 221:129–143.Medline, CAS, Google Scholar
- 12. Matz, M. V. 2003. Amplificazione di pool di cDNA rappresentativi da quantità microscopiche di tessuto animale. Metodi Mol. Biol. 221:103–116.Medline, CAS, Google Scholar
- 13. Il sito utilizza cookie tecnici e cookie di profilazione di terze parti. 2003. Amplificazione globale di mRNA mediante template-switching PCR: linearità e applicazione all’analisi di microarray. Acidi nucleici Res. 22:e142.Crossref, Google Scholar
- 14. Polacek, DC, A. G. Passerini, C. Shi, N. M. di Francesco, E. Manduchi, G. R. Grant, S. Powell, H. Bischof, et al.. 2003. Fedeltà e maggiore sensibilità dei profili di trascrizione differenziali a seguito di amplificazione lineare di quantità nanogram di mRNA endoteliale. Physiol. Genomica 13:147-156.Crossref, Medline, CAS, Google Scholar
- 15. Wilson, C., S. D. Pepper, Y. Hey, e C. J. Miller. 2004. I protocolli di amplificazione introducono errori sistematici ma riproducibili negli studi di espressione genica. BioTechniques 36: 498-506.Link, CAS, Google Scholar
- 16. Ohara, O., T. Nagase, G. Il suo nome deriva dal latino “H. Kohga”, “R. Kikuno”, “S. Hiraoka”, “Y. Takahashi”, “S. Kitajima”, et al.. 2002. Caratterizzazione della libreria cDNA frazionata in dimensione generata dal metodo in vitro recombination-assisted. DNA Res. 9: 47-57.Crossref, Medline, CAS, Google Scholar
- 17. Ohara, O. 2003. Costruzione di una libreria di cDNA frazionata a dimensione assistita da una reazione di ricombinazione in vitro. Metodi Mol. Biol. 221:59–71.Medline, CAS, Google Scholar
- 18. Gubler, U. e B. J. Hoffman. 1983. Un metodo semplice e molto efficiente per la generazione di librerie cDNA. Gene 25:263-269.Crossref, Medline, CAS, Google Scholar
- 19. Sambrook, J. e D. W. Russell. 2001. Preparazione del DNA plasmidico mediante lisi alcalina con SDS, p. 1.31-1.42. Clonazione molecolare, (3a edizione). CSH Laboratory Press, Cold Spring Harbor, NY.Google Scholar
- 20. Chiesa, G. M. e W. Gilbert. 1984. Sequenziamento genomico. Proc. Natl. Acad. Sic. USA 81:1991-1995.Crossref, Medline, CAS, Google Scholar
- 21. Il sito utilizza cookie tecnici e di terze parti. 2000. Modelli di utilizzo del segnale di poliadenilazione variante nei geni umani. Genoma Res. 10:1001-1010.Crossref, Medline, CAS, Google Scholar
- 22. Il suo nome deriva dal latino “Tanaka”, “T. S.”, “S. A. Jaradat”, “M. K. Lim”, “G. J. Kargul”, “X. Wang”, “M. J. Grahovac”, “S. Pantano”, “Y. Sano”, et al.. 2000. Genome-wide espressione profiling di mid-gestazione placenta e embrione utilizzando un 15.000 mouse developmental cDNA microarray. Proc. Natl. Acad. Sic. Stati Uniti D’America 97: 9127-9132.Crossref, Medline, Google Scholar
- 23. Lukyanov, K., L. Diatchenko, A. Chenchik, A. Nanisetti, P. Siebert, N. Usman, M. Matz e S. Lukyanov. 1997. Costruzione di librerie cDNA da piccole quantità di RNA totale utilizzando l’effetto PCR di supressione. Biochimica. Biophys. Res. Commun. 230:285–288.Crossref, Medline, CAS, Google Scholar
- 24. Il suo nome deriva dal nome della città. 2001. Costruzione di librerie cDNA arricchite con trascrizioni lunghe da quantità submicrogram di RNA totali mediante un metodo di amplificazione PCR universale. Genoma Res. 11:1553-1558.Crossref, Medline, CAS, Google Scholar