
Perché sviluppare l’analisi Cap dell’espressione genica?
La ricerca di Piero Carninci e Yoshihide Hayashizaki alla fine degli anni 1990, iniziata con il metodo cap trapper, l’uso del trealosio, il metodo di normalizzazione / sottrazione e un nuovo vettore di clonazione, ha posto le basi per lo sviluppo dell’analisi Cap dell’espressione genica. Con cap trapper, gli ibridi cDNA/mRNA a lunghezza intera sono isolati e l’mRNA è chimicamente biotinilato sulla struttura del cappuccio e le perle magnetiche rivestite con streptavidina catturano gli ibridi. Questo progresso nella serie delle principali tecnologie del DNA è descritto dalla Natura come Pietra Miliare 5 (www.nature.com/milestones/miledna/full/miledna05.html). Il loro scopo nello sviluppo di CAGE era quello di creare una tecnologia per mappare in modo completo la stragrande maggioranza dei siti di partenza della trascrizione umana e dei loro promotori. Infatti, la tecnologia per profilare l’attività di trascrizione genica in ogni sito promotore non esisteva fino CAGE è venuto sulla scena. L’RNA messaggero (mRNA) rappresenta un legame critico tra le informazioni codificate nei singoli geni su un genoma e la composizione proteica che determina il destino di un organismo. Ci siamo chiesti: quali sono le regioni genomiche, o promotori, che guidano l’espressione specifica dei geni e dei loro RNA univocamente diversi? In effetti, le raccolte di cDNA a lunghezza intera hanno dimostrato che la maggior parte dei geni ha più di un sito di partenza della trascrizione e quindi è abbastanza difficile identificare le regioni di controllo, cioè i promotori, responsabili dell’espressione delle varie forme di trascrizioni.
Analizzare la complessità di questo processo di trasferimento delle informazioni, chiamato ‘trascrizione’, richiede lo sviluppo di sofisticati strumenti molecolari in grado di catturare sia gli aspetti qualitativi che quantitativi dell’espressione genica. Quindi, con CAGE, la nostra tecnologia di rilevamento iniziale della trascrizione a livello genomico originale, possiamo fare un profilo di espressione genica ad alta intensità con l’identificazione simultanea dei siti di partenza trascrizionali specifici del tessuto/cellula/condizione (TSS), inclusa l’analisi dell’utilizzo del promotore. CAGE si basa sulla preparazione e sequenziamento di concatameri di DNA tag derivanti dai 20 nucleotidi iniziali da 5′ end mRNA, che riflettono la concentrazione originale di mRNA nel campione analizzato (frequenza di RNA).
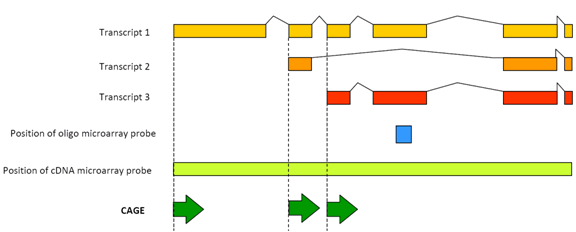
Figura 1: CAGE rileva l’attività trascrizionale di ogni trascrizione del promotore.
I tag Expressed Sequencing (EST) sono stati utilizzati nelle prime fasi dello sviluppo tecnologico per identificare gli elementi promotori allineandoli sul genoma umano. Tuttavia, questo processo è molto costoso a causa dei costi di gestione dei CDNA fisici e del sequenziamento Sanger. Un modo per superare questi problemi è utilizzare tecnologie di tagging, che sono state sviluppate per rilevare trascrizioni con una sensibilità che è almeno un ordine di grandezza maggiore del sequenziamento EST, quindi identificare esaustivamente trascrizioni, identificare i loro promotori e correlarli con i profili di espressione contando i tag come misura digitale dell’espressione genica.
Queste sequenze (chiamate anche “tag”) vengono quindi allineate alle sequenze del genoma mediante semplici procedure computazionali (chiamate BLAST) e contate, il che fornisce una misurazione della frequenza di espressione dell’RNA. Poiché questi tag di sequenza identificano i siti di partenza della trascrizione dell’RNA, identificano anche le sequenze del genoma vicino a quei siti di partenza. Le regioni limitrofe sono le regioni principali del promotore, che sono sequenze genomiche che causano la trascrizione dei geni nelle molte condizioni diverse che si incontrano in molti organismi complessi, dai topi agli esseri umani.
Attributi di CAGE
CAGE presenta grandi vantaggi rispetto alle classiche tecniche di rilevamento delle espressioni basate su microarray. Infatti, identificando il promotore che causa la trascrizione dell’RNA in ogni fenomeno biologico, tessuto, cellula ecc. possiamo identificare gli elementi regolatori del DNA specifici per ogni fenomeno biologico osservando le sequenze che si trovano nei promotori delle isoforme di RNA espresse nei campioni analizzati. I promotori contengono sequenze specifiche, o siti di legame del fattore di trascrizione (TFBS), che sono riconosciuti dalle loro proteine leganti chiamate fattori di trascrizione (TF) e promuovono, o in alternativa reprimono, la trascrizione. Usando metodi computazionali, i nostri ricercatori analizzano i promotori con profili di espressione simili per i loro TFBS e quindi identificano i TFS responsabili dell’output trascrizionale del genoma. Contando il numero di tag di GABBIA per ciascun promotore all’interno di un gene, possiamo ora determinare non solo il livello di espressione dell’RNA (questo è un rilevamento digitale della frequenza) ma, soprattutto, anche da quale dei vari promotori alternativi l’RNA viene trascritto.
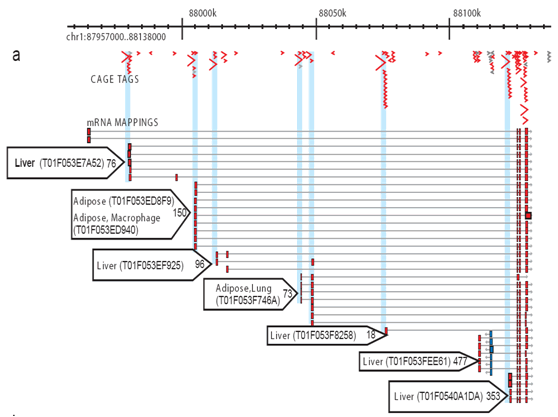
Figura 2: CAGE consente la profilazione completa degli attivi in ogni sito promotore. Per ogni libreria, un certo numero di tag GABBIA sono sequenziati e allineati al genoma in modo che la specifica attività trascrizionale a ciascun promotore può essere misurata e il contributo di ogni promotore distinto. Questo esempio semplificato mostra i promotori del nucleo adiposo e epatico. Minuscole frecce blu: tag GABBIA individuale; frecce rosse: preferenza di utilizzo del promotore per i tessuti; scatole rosse: regioni del promotore principale.
Come accennato, CAGE utilizza il cap-trapping come primo passo per catturare le estremità 5′ dei CDNA, che vengono poi trasformate in brevi sequenze (tag) da 20 a 27 nt corrispondenti all’mRNA TSSs, . Abbiamo prodotto milioni di tag GABBIA per topi e umani usando tag GABBIA concatenati con sequenziamento Sanger, fino a quando recentemente ci siamo trasferiti a deepCAGE, per il quale usiamo sequenziamento di seconda generazione.
Fino al 2006 gestivamo librerie a GABBIA sulla nostra pipeline di sequenziamento RISA originale, che è stata costruita alla fine degli anni ‘ 90 e comprendeva l’unico sequencer capillare con un array di 384 capillari che abbiamo sviluppato in collaborazione con la Shimadzu Corporation.
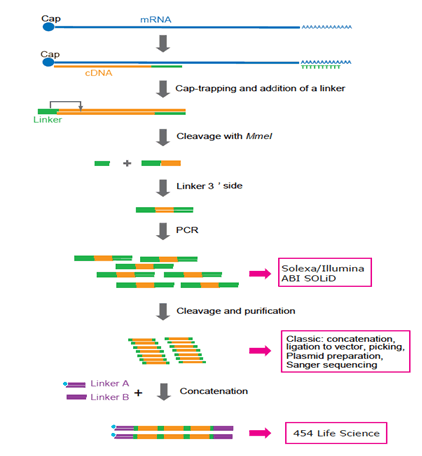
Figura 3: Rappresentazione del protocollo di preparazione della GABBIA adattato a varie piattaforme. Ora Solexa e Illumina sono preferiti. 454 Life Sciences (sistema FLX) non viene più utilizzato perché la concatenazione richiede cicli di PCR aggiuntivi e manipolazioni complicate. In futuro, la tecnologia di sequenziamento a singola molecola sarà preferita perché la PCR potrebbe non essere richiesta.
Sebbene la tecnologia di tagging e sequenziamento sia stata sviluppata utilizzando il metodo serial analysis of gene analysis (SAGE), CAGE è unica perché si basa sul principio di profilare l’estremità 5′ degli RNA che trasportano un MRNA cap-site – all e una grande frazione degli RNA non codificanti. Abbiamo sviluppato tag a GABBIA lunga 27-nt per aumentare l’efficienza di mappatura.