
miért kell fejleszteni a génexpresszió Cap elemzését?
Piero Carninci és Yoshihide Hayashizaki kutatása az 1990-es évek végén, amely a cap trapper módszerrel, a trehalóz használatával, a normalizálási/kivonási módszerrel és egy új klónozási vektorral kezdődött, megteremtette a terepet a génexpresszió Cap elemzésének kidolgozásához. A cap trapper segítségével teljes hosszúságú cDNS/mRNS hibrideket izolálnak, és az mRNS-t kémiailag biotinilezik a sapka szerkezetén, és a sztreptavidinnel bevont mágneses gyöngyök elfogják a hibrideket. Ezt az előrelépést a főbb DNS-technológiák sorozatában a természet az 5. mérföldkőnek nevezi (www.nature.com/milestones/miledna/full/miledna05.html). a CAGE fejlesztésében az volt a céljuk, hogy olyan technológiát hozzanak létre, amely átfogóan feltérképezi az emberi transzkripciós kiindulási helyek túlnyomó többségét és promótereiket. Valójában a gén transzkripció aktivitásának profilozására szolgáló technológia az egyes promóter helyeken nem létezett, amíg CAGE a helyszínre nem került. A Messenger RNS (mRNS) kritikus kapcsolatot jelent a genom egyes génjeiben kódolt információ és a szervezet sorsát meghatározó fehérje összetétel között. Megkérdeztük magunktól: melyek azok a genomiális régiók vagy promóterek, amelyek a gének specifikus expresszióját és egyedülállóan különböző RNS-ét vezérlik? Valójában a teljes hosszúságú cDNS-gyűjtemények kimutatták, hogy a legtöbb génnek egynél több transzkripciós kiindulási helye van, ezért meglehetősen nehéz azonosítani a transzkripciók különböző formáinak expressziójáért felelős szabályozó régiókat, azaz promotereket.
A transzkripciónak nevezett információátviteli folyamat összetettségének elemzése olyan kifinomult molekuláris eszközök kifejlesztését igényli, amelyek képesek megragadni a génexpresszió kvalitatív és kvantitatív aspektusait is. Tehát a cage-vel, az eredeti Genom egészére kiterjedő transzkripciós kezdő detektálási technológiánkkal, magas szintű génexpressziós profilozást végezhetünk a szövet/sejt/állapot specifikus transzkripciós kiindulási helyek (TSS) egyidejű azonosításával, beleértve a promoter használat elemzését is. A CAGE a kezdeti 20 nukleotidból származó DNS-címkék előkészítésén és szekvenálásán alapul 5 ‘ end mRNS-ek, amelyek tükrözik az mRNS eredeti koncentrációját az elemzett mintában (RNS frekvencia).
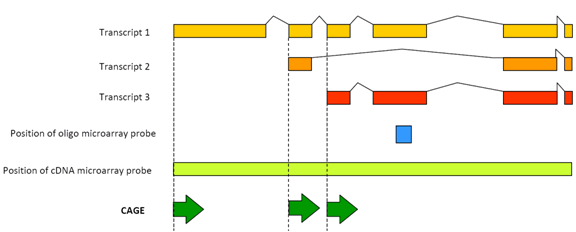
1.ábra: A Cage felismeri az egyes promoter átiratok transzkripciós aktivitását.
expresszált szekvenáló címkéket (est) használtunk a technológia fejlődésének korai szakaszában a promoter elemek azonosítására az emberi genomhoz való igazítással. Azonban ez a folyamat nagyon drága, mert a költségek kezelése fizikai cDNS és Sanger szekvenálás. E problémák leküzdésének egyik módja a címkézési technológiák használata, amelyeket az EST szekvenálásnál legalább egy nagyságrenddel nagyobb érzékenységű átiratok kimutatására fejlesztettek ki, majd kimerítően azonosítják az átiratokat, azonosítják promótereiket és korrelálják őket az expressziós profilokkal azáltal, hogy a címkéket a génexpresszió digitális mércéjeként számolják.
ezeket a szekvenciákat (más néven “címkék”) ezután egyszerű számítási eljárásokkal (BLAST) igazítják a genomszekvenciákhoz, és megszámolják, ami az RNS-expresszió gyakoriságának mérését adja. Mivel ezek a szekvenciacímkék azonosítják az RNS transzkripció kiindulási helyeit, azonosítják a kiindulási helyekhez közeli genomszekvenciákat is. A szomszédos régiók a mag promoter régiók, amelyek olyan genomi szekvenciák, amelyek a gének átírását okozzák a sokféle körülmények között, amelyek sok összetett organizmusban előfordulnak, egerektől emberig.
A CAGE attribútumai
A CAGE nagy előnyökkel rendelkezik a klasszikus mikroarray-alapú expressziós detektálási technikákhoz képest. Valójában az RNS transzkripciót okozó promoter azonosításával minden biológiai jelenségben, szövetben, sejtben stb. azonosíthatjuk az egyes biológiai jelenségekre specifikus DNS-szabályozó elemeket, ha megnézzük azokat a szekvenciákat, amelyek az elemzett mintákban expresszált RNS-izoformák promotereiben vannak. A promoterek specifikus szekvenciákat vagy transzkripciós faktor kötőhelyeket (TFB-ket) tartalmaznak, amelyeket a transzkripciós faktoroknak (TF) nevezett kötő fehérjéik felismernek, és elősegítik, vagy alternatív módon elnyomják a transzkripciót. Számítási módszerekkel, kutatóink elemzik a hasonló expressziós profilokkal rendelkező promótereket TFB-jükhöz, majd azonosítják a genom transzkripciós kimenetéért felelős TF-eket. A génen belüli egyes promoterek ketrec-címkéinek megszámlálásával most már nemcsak az RNS expressziós szintjét határozhatjuk meg (ez a frekvencia digitális detektálása), hanem fontos, hogy a különféle alternatív promoterek közül melyikből írják át az RNS-t.
2.ábra: a ketrec lehetővé teszi az aktiválások átfogó profilozását minden promoter helyszínen. Minden könyvtárhoz számos ketrec-címkét szekvenálnak és igazítanak a genomhoz, így az egyes promóterek specifikus transzkripciós aktivitása mérhető, és megkülönböztethető az egyes promóterek hozzájárulása. Ez az egyszerűsített példa a zsír-és májmag-promotereket mutatja. Apró kék nyilak: egyedi ketrec címkék; piros nyilak: promoter Használati preferencia szövetek; piros dobozok: core promoter régiók.
mint említettük, a CAGE a cap-csapdázást használja az első lépésként a cDNS-ek 5′ végeinek rögzítésére, amelyeket ezután 20-27 nt rövid szekvenciává (címkékké) alakítanak át, amelyek megfelelnek az mRNS TSSs-nek, . Több millió egér és emberi KETRECCÍMKÉT állítottunk elő összefűzött Ketreccímkékkel Sanger szekvenálással, amíg nemrégiben át nem költöztünk a deepCAGE-be, amelyhez második generációs szekvenálást használunk.
2006-ig KETRECKÖNYVTÁRAKAT működtettünk az eredeti RISA szekvenáló csővezetékünkön, amelyet a 90-es évek végén építettek, és tartalmazta az egyetlen kapilláris szekvenszert 384 kapilláris tömbbel, amelyet a Shimadzu Corporation-vel együttműködve fejlesztettünk ki.
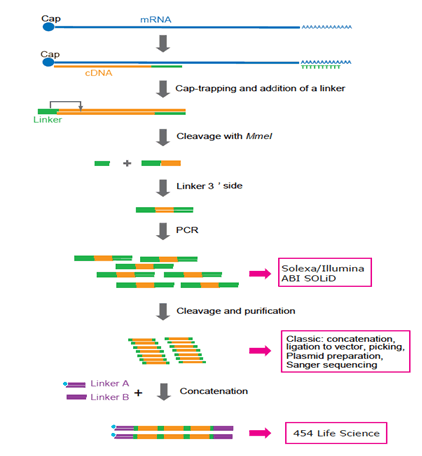
3.ábra: a KETRECKÉSZÍTÉSI protokoll ábrázolása különböző platformokhoz igazítva. Most Solexa és Illumina előnyben részesülnek. A 454 Life Sciences (FLX system) rendszert már nem használják, mert az összefűzés további PCR-ciklusokat és bonyolult manipulációt igényel. A jövőben az egymolekulás szekvenálási technológiát részesítik előnyben, mert PCR nem feltétlenül szükséges.
bár a címkézési és szekvenálási technológiát a serial analysis of gene analysis (Sage) módszerrel fejlesztették ki, a CAGE egyedülálló, mivel azon az elven alapul, hogy az RNS-ek 5′ végét profilozzák, amely egy cap – site-all mRNS-t és a nem kódoló RNS-ek nagy részét hordozza. Az általunk kifejlesztett 27-nt hosszú ketrec címkék, hogy növelje a leképezés hatékonyságát.