
miksi kehittää geeniekspression Cap-analyysia?
Piero Carnincin ja Yoshihide Hayashizakin 1990-luvun lopulla tekemä tutkimus, joka alkoi cap trapper-menetelmällä, trehaloosin käytöllä, normalisointi – / vähennyslaskumenetelmällä ja uudella kloonausvektorilla, asetti vaiheen geenien ekspression Cap-analyysin kehittämiselle. Cap trapperin avulla eristetään täyspitkät cDNA/mRNA-hybridit, ja mRNA biotinyloidaan kemiallisesti cap-rakenteeseen ja streptavidiinipäällysteiset magneettihelmet sieppaavat hybridit. Tämä eteneminen suurten DNA-teknologioiden sarjassa on luonnehdittu virstanpylvääksi 5 (www.nature.com/milestones/miledna/full/miledna05.html heidän tarkoituksenaan Cagen kehittämisessä oli luoda teknologia, jolla voidaan kartoittaa kattavasti valtaosa ihmisten transkription aloituspaikoista ja niiden promoottoreista. Itse asiassa teknologia, joka profiloi geenien transkription toiminnan promoottoripaikoilla, ei ollut olemassa ennen kuin CAGE tuli paikalle. Lähetti-RNA (mRNA) edustaa kriittistä yhteyttä genomin yksittäisiin geeneihin koodatun tiedon ja organismin kohtaloa määrittävän proteiinimeikin välillä. Kysyimme itseltämme: mitkä ovat genomialueet tai promoottorit, jotka ohjaavat geenien ja niiden ainutlaatuisen erilaisten RNA: iden erityistä ilmentymistä? Täyspitkät cDNA-kokoelmat ovatkin osoittaneet, että useimmilla geeneillä on useampi kuin yksi transkription aloituspaikka, joten on melko vaikeaa tunnistaa kontrollialueita eli promoottoreita, jotka vastaavat transkriptioiden eri muotojen ilmentymisestä.
tämän ”transkriptioksi” kutsutun tiedonsiirtoprosessin monimutkaisuuden analysointi edellyttää kehittyneiden molekyylityökalujen kehittämistä, joilla voidaan kuvata sekä geenien ilmentymisen laadullisia että määrällisiä näkökohtia. Joten CAGE, alkuperäinen genomin laajuinen transkriptio alkaa tunnistus tekniikka, voimme tehdä high-through geeniekspression profilointi samanaikaisella tunnistuksella kudoksen / solun / tilan spesifisen transkription start sites (TSS), mukaan lukien promoottori käyttöanalyysi. CAGE perustuu valmistamiseen ja sekvensointiin DNA-tageja, jotka johtuvat 20 nukleotidia 5 ’ end mRNAs, jotka heijastavat alkuperäisen pitoisuus mRNA analysoitu näyte (RNA taajuus).
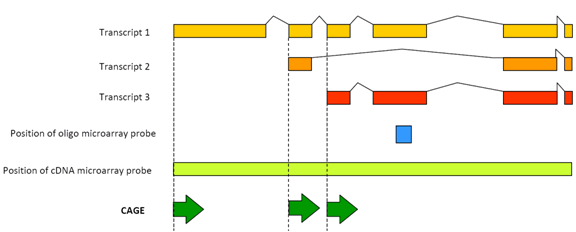
Kuva 1: CAGE havaitsee jokaisen promoottorin transkription aktiivisuuden.
Expressed sequencing tageja (ests) käytettiin teknologian kehityksen alkuvaiheessa promoottorielementtien tunnistamiseen kohdistamalla ne ihmisen genomiin. Tämä prosessi on kuitenkin erittäin kallis fyysisten cdnojen ja Sanger-sekvensoinnin käsittelykustannusten vuoksi. Yksi tapa ratkaista nämä ongelmat on käyttää merkintätekniikoita, jotka on kehitetty havaitsemaan transkriptit, joiden herkkyys on vähintään yhtä suuruusluokkaa suurempi kuin est-sekvensointi, sitten tyhjentävästi tunnistaa transkriptit, tunnistaa niiden promoottorit ja korreloida ne ekspressioprofiilien kanssa laskemalla tunnisteet geeniekspression digitaalisena mittana.
nämä sekvenssit (joita kutsutaan myös ”tägeiksi”) sovitetaan sitten genomisekvensseihin yksinkertaisilla laskennallisilla toimenpiteillä (joita kutsutaan BLASTIKSI) ja lasketaan, jolloin saadaan RNA-ekspression taajuuden mittaus. Koska nämä sekvenssitagit tunnistavat RNA-transkription aloituspaikat, ne tunnistavat myös genomisekvenssit, jotka ovat lähellä aloituspaikkoja. Lähialueet ovat ydinpromoottorialueita, jotka ovat genomisekvenssejä, jotka aiheuttavat geenien litteroitumista monissa erilaisissa olosuhteissa, joita esiintyy monissa monimutkaisissa eliöissä hiiristä ihmisiin.
häkin attribuuteilla
häkillä on suuria etuja verrattuna klassisiin microarray-pohjaisiin lausekkeen ilmaisutekniikoihin. Itse asiassa tunnistamalla promoottori, joka aiheuttaa RNA-transkription jokaisessa biologisessa ilmiössä, kudoksessa, solussa jne. voimme tunnistaa DNA: n säätelyelementit, jotka ovat spesifisiä kullekin biologiselle ilmiölle tarkastelemalla sekvenssejä, jotka ovat RNA-isoformien promoottoreissa ilmaistuna analysoiduissa näytteissä. Promoottorit sisältävät erityisiä sekvenssejä eli transkriptiotekijän sitoutumispaikkoja (tfbs), jotka tunnistetaan niiden sitoutumisproteiineista, joita kutsutaan transkriptiotekijöiksi (TF) ja jotka edistävät tai vaihtoehtoisesti tukahduttavat transkriptiota. Laskennallisten menetelmien avulla tutkijamme analysoivat promoottoreita, joilla on samanlaiset ekspressioprofiilit tfbs: lle ja sitten tunnistavat TFS: n, joka vastaa genomin transkriptiotuotoksesta. Laskemalla kunkin geenin sisällä olevan promoottorin HÄKKILAPPUJEN määrän voimme nyt määrittää paitsi RNA: n ekspressiotason (tämä on taajuushavainto digitaalisesti) myös, mikä on tärkeää, myös sen, mistä eri vaihtoehtoisista promoottoreista RNA on transkriboitu.
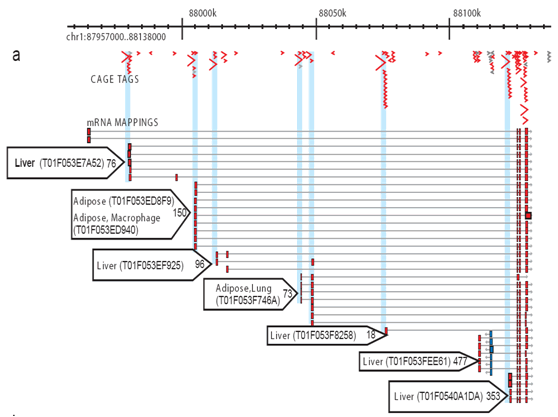
kuva 2: CAGE mahdollistaa aktivoijien kattavan profiloinnin jokaisessa promoottoripaikassa. Kutakin kirjastoa varten sekvensoidaan useita HÄKKILAPPUJA, jotka ovat linjassa genomin kanssa, jotta kunkin promoottorin spesifinen transkriptiotoiminta voidaan mitata ja kunkin promoottorin osuus erottaa toisistaan. Tässä yksinkertaistetussa esimerkissä esitetään rasva – ja maksaytimien promoottorit. Tiny blue arrows: yksittäiset häkki tunnisteet; punaiset nuolet: promoottori käyttö mieltymys kudosten; punaiset laatikot: core promoottori alueet.
kuten edellä mainittiin, CAGE käyttää cap-ansastusta ensimmäisenä askeleena pyydystääkseen cDNAs: N 5′ – päät, jotka sitten muunnetaan lyhyeksi jaksoksi (tagit) 20-27 nt vastaa mRNA TSS: ää . Olemme tuottaneet miljoonia hiiren ja ihmisen HÄKKILAPPUJA käyttäen Sanger-sekvensoinnilla varustettuja häkkilappuja, kunnes siirryimme hiljattain deepcageen, jossa käytämme toisen sukupolven sekvensointia.
vuoteen 2006 asti pyöritimme CAGE-kirjastoja alkuperäisellä RISA-sekvensointiputkellamme, joka rakennettiin 90-luvun lopulla ja sisälsi ainoan kapillaarisekvensserin, jossa oli 384 kapillaaria, jotka kehitimme yhteistyössä Shimadzu Corporationin kanssa.
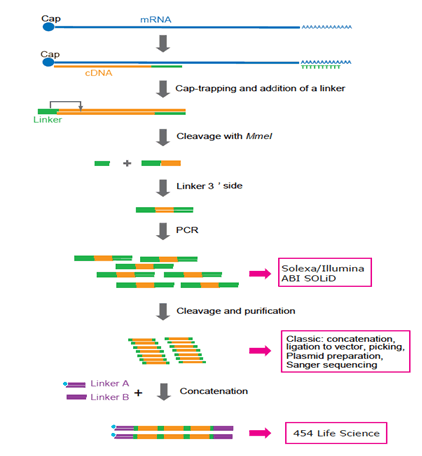
kuva 3: esitys eri alustoille sovitetusta häkkien valmisteluprotokollasta. Nyt suositaan Solexaa ja Illuminaa. 454 biotieteitä (FLX-järjestelmä) ei käytetä enää, koska tiivistäminen vaatii lisää PCR-syklejä ja monimutkaista manipulointia. Tulevaisuudessa suositaan yhden molekyylin sekvensointitekniikkaa, koska PCR: ää ei välttämättä tarvita.
vaikka tunnistus-ja sekvensointiteknologia on kehitetty käyttämällä serial analysis of gene analysis (SAGE) – menetelmää, CAGE on ainutlaatuinen, koska se perustuu periaatteeseen profiloida 5′-Pää RNAs, jossa on cap-site-kaikki mrnat ja suuri osa ei-koodaa RNAs. Olemme kehittäneet 27-nt pitkä häkki tunnisteet lisätä kartoituksen tehokkuutta.