
¿Por qué desarrollar el Análisis de Cap de la Expresión Génica?
La investigación de Piero Carninci y Yoshihide Hayashizaki a finales de la década de 1990, que comenzó con el método cap trapper, el uso de trehalosa, el método de normalización/sustracción y un nuevo vector de clonación, sentó las bases para el desarrollo del Análisis Cap de la Expresión Génica. Con cap trapper, se aíslan híbridos de cDNA/ARNm de longitud completa, y el ARNm se biotinila químicamente en la estructura de la tapa y las perlas magnéticas recubiertas de estreptavidina capturan a los híbridos. Este avance en la serie de las principales tecnologías de ADN es descrito por la Naturaleza como Hito 5 (www.nature.com/milestones/miledna/full/miledna05.html Su propósito en el desarrollo de CAGE fue crear una tecnología para mapear exhaustivamente la gran mayoría de los sitios de inicio de transcripción humana y sus promotores. De hecho, la tecnología para perfilar la actividad de la transcripción de genes en cada sitio promotor no existía hasta que CAGE entró en escena. El ARN mensajero (ARNm) representa un vínculo crítico entre la información codificada en los genes individuales de un genoma y la composición proteica que determina el destino de un organismo. Nos preguntamos: ¿cuáles son las regiones genómicas, o promotores, que impulsan la expresión específica de genes y sus ARN singularmente diferentes? De hecho, las colecciones completas de ADNc han demostrado que la mayoría de los genes tienen más de un sitio de inicio de transcripción y, por lo tanto, es bastante difícil identificar las regiones controladoras, es decir, los promotores, responsables de la expresión de las diversas formas de transcripciones. Analizar la complejidad de este proceso de transferencia de información, llamado ‘transcripción’, requiere el desarrollo de herramientas moleculares sofisticadas que puedan capturar los aspectos cualitativos y cuantitativos de la expresión génica. Por lo tanto, con CAGE, nuestra tecnología original de detección de inicio de transcripción de todo el genoma, podemos realizar perfiles de expresión génica de alto nivel con identificación simultánea de los sitios de inicio de transcripción (SST) específicos de tejido/célula/condición, incluido el análisis del uso del promotor. CAGE se basa en la preparación y secuenciación de concatámeros de etiquetas de ADN derivadas de los 20 nucleótidos iniciales de ARNm de extremo 5′, que reflejan la concentración original de ARNm en la muestra analizada (frecuencia de ARN).
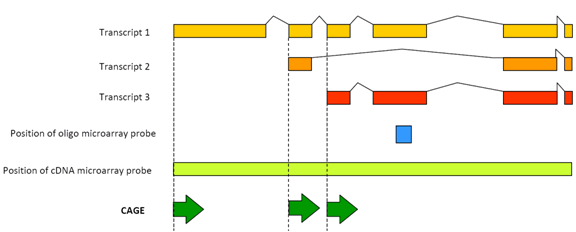
Figura 1: CAGE detecta la actividad transcripcional de cada transcripción de promotor.
Las etiquetas de secuenciación expresa (EST) se utilizaron en las primeras etapas del desarrollo tecnológico para identificar elementos promotores alineándolos con el genoma humano. Sin embargo, este proceso es muy costoso debido a los costos de manejar cDNAs físicos y secuenciación de Sanger. Una forma de superar estos problemas es utilizar tecnologías de etiquetado, que se han desarrollado para detectar transcripciones con una sensibilidad que es al menos un orden de magnitud mayor que la secuenciación EST, luego identificar exhaustivamente las transcripciones, identificar a sus promotores y correlacionarlos con perfiles de expresión contando las etiquetas como una medida digital de la expresión génica.
Estas secuencias (también llamadas «etiquetas») se alinean con secuencias genómicas mediante procedimientos computacionales simples (llamados BLAST) y se cuentan, lo que da una medición de la frecuencia de expresión de ARN. A medida que estas etiquetas de secuencia identifican los sitios de inicio de la transcripción de ARN, también identifican las secuencias del genoma cercanas a esos sitios de inicio. Las regiones vecinas son las regiones promotoras centrales, que son secuencias genómicas que hacen que los genes se transcriban en las muchas condiciones diferentes que se encuentran en muchos organismos complejos, desde ratones hasta humanos.
Atributos de la JAULA
LA JAULA tiene grandes ventajas en comparación con las técnicas clásicas de detección de expresión basadas en microarrays. De hecho, al identificar el promotor que causa la transcripción de ARN en cada fenómeno biológico, tejido, célula, etc. podemos identificar los elementos reguladores del ADN que son específicos para cada fenómeno biológico observando las secuencias que se encuentran en los promotores de las isoformas de ARN que se expresan en las muestras analizadas. Los promotores contienen secuencias específicas, o sitios de unión de factores de transcripción (TFB), que son reconocidos por sus proteínas de unión llamadas factores de transcripción (TF) y promueven, o alternativamente reprimen, la transcripción. Utilizando métodos computacionales, nuestros investigadores analizan promotores que tienen perfiles de expresión similares para sus TFBS y luego identifican los TFs responsables de la salida transcripcional del genoma. Al contar el número de etiquetas de JAULA para cada promotor dentro de un gen, ahora podemos determinar no solo el nivel de expresión de ARN (esto es una detección digital de frecuencia), sino, lo que es más importante, también de cuál de los diversos promotores alternativos se transcribe el ARN.
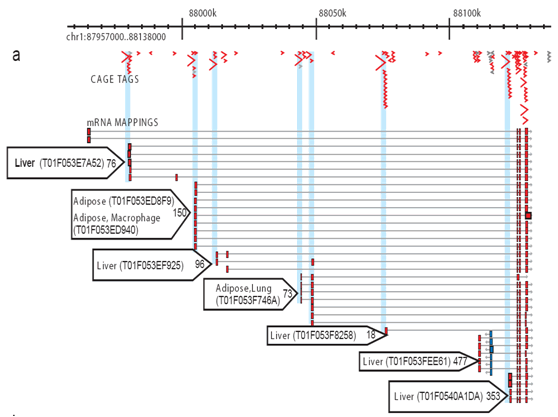
Figura 2: CAGE permite el perfil completo de activaciones en cada sitio del promotor. Para cada biblioteca, se secuencian varias etiquetas de JAULA y se alinean con el genoma para que se pueda medir la actividad transcripcional específica de cada promotor y distinguir la contribución de cada promotor. Este ejemplo simplificado muestra promotores del núcleo adiposo y hepático. Pequeñas flechas azules: etiquetas de JAULA individuales; flechas rojas: preferencia de uso del promotor para los tejidos; cajas rojas: regiones del promotor del núcleo.
Como se mencionó, CAGE utiliza la captura de cap como primer paso para capturar los extremos de 5′ de los cDNAs, que luego se transforman en una secuencia corta (etiquetas) de 20 a 27 nt correspondientes al ARNm TSSs, . Hemos producido millones de etiquetas de JAULA para ratones y humanos utilizando etiquetas de JAULA concatenadas con secuenciación Sanger, hasta que recientemente nos mudamos a deepCAGE, para lo cual usamos secuenciación de segunda generación.
Hasta 2006, estábamos ejecutando bibliotecas de JAULAS en nuestro pipeline de secuenciación RISA original, que se construyó a finales de los 90 e incluía el único secuenciador capilar con una matriz de 384 capilares que desarrollamos en colaboración con Shimadzu Corporation.
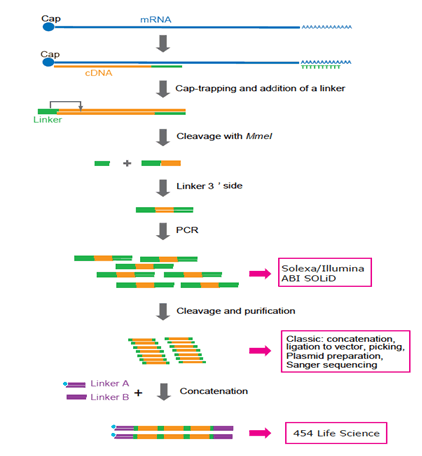
Figura 3: Representación del protocolo de preparación de JAULAS adaptado a varias plataformas. Ahora Solexa e Illumina son preferidas. 454 Life Sciences (sistema FLX) ya no se utiliza porque la concatenación requiere ciclos de PCR adicionales y una manipulación complicada. En el futuro, se preferirá la tecnología de secuenciación de una sola molécula porque es posible que no se requiera PCR.
Aunque la tecnología de etiquetado y secuenciación se ha desarrollado utilizando el método de análisis en serie de análisis de genes (SAGE), CAGE es única porque se basa en el principio de perfilar el extremo 5′ de los ARN que llevan un ARNm de todo el sitio de la tapa y una gran fracción de los ARN no codificantes. Hemos desarrollado etiquetas DE JAULA larga de 27 nt para aumentar la eficiencia del mapeo.