
Una parte importante de la comprensión de cómo funcionan estos sistemas para controlar la división celular fue el descubrimiento de que el p53 afecta tanto al cáncer como al envejecimiento. Tyner et al. (2002) idearon una estrategia agenética en ratones para comparar los efectos de la ausencia de proteína p53 o más pequeña que la proteína p53 normal. Las dos líneas de ratones transgénicos mutantes tenían una deleción completa del gen p53 (p53 -) o una forma truncada de p53 (p53m, mutante) que no tenía los primeros seis exones del gen p53 (Figura 2).
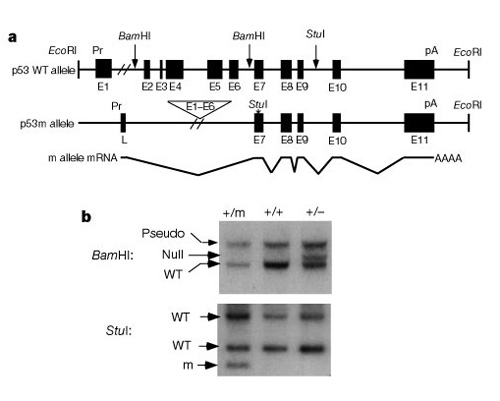
La primera mitad del estudio comparó tres grupos de ratones:grupo 1, p53+ / p53-(eliminación de una copia de p53); grupo 2, p53+ / p53m(mutante de eliminación parcial); y grupo 3, p53+ / p53 normal). La tabla 1 muestra estos tres grupos y sus resultados experimentales relacionados con los fenotipos de cáncer y envejecimiento. Curiosamente, ninguno de los ratones del grupo 2, con la proteína p53 truncada, desarrolló tumores potencialmente mortales, mientras que el 45% del grupo 3 (tipo salvaje) y más del 80% del grupo 1 desarrollaron tumores potencialmente mortales. Los ratones del grupo 2 también tuvieron una vida media intermedia entre la vida muy corta del grupo 1 y la vida más larga de los ratones de tipo salvaje del grupo 3. La conclusión de estos datos es que la mutación parcial p53 redujo la incidencia del cáncer y, al mismo tiempo, pareció causar un déficit en la esperanza de vida, no una prolongación de la vida.
| la Tabla 1. Experimental results from genetic mousestudies with p53 mutants | |||
| Genotype | Cancer phenotype | Agingphenotype | |
| Group 1 | p53+/p53- (complete deletion) | 80%had tumors | Muchshorter life span |
| Group 2 | p53+/p53m (partial mutant) | None | Shorterlife span |
| Group 3 | p53+/p53+ (wild type) | 45%had tumors | Normallife span |
| Adaptado de Tyner etal. 2002 | |||
Los autores también señalaron que los ratones del grupo 2 desarrollaron fenotipos característicos de ratones viejos, como crecimiento lento del pelo y espinas jorobadas debido a cambios esqueléticos, antes de lo que hacían los ratones salvajes (Figura 3).

En la segunda mitad del estudio, Tyner y colleaguesasked si el p53 mutantfunctioned de manera diferente en la presencia de la normal de p53. Criaron una línea adicional de ratones transgénicos con lelos mutantes p53- / p53m y descubrieron que estos ratones no tenían la fuerte protección tumoral y mostraban un efecto de vida mucho menor. Así que el mutante p53 de alguna manera necesitaba trabajar inconcluso con el p53 normal para tener un efecto. Como observación general, los investigadores informaron que las células del heterocigoto p53+ / p53m resultaron ser aproximadamente tres veces más duras que las células de tipo salvaje. Por lo tanto, aunque estas células eran resistentes al cáncer, este fondo trasero p53+/ p53 también causó un envejecimiento más temprano. De hecho, la actividad de p53 en estos termocigotos parecía ser notablemente más alta que su actividad en la naturaleza type.It parecía que tal cambio, aunque a priori, sería bueno contra el cáncer y la senescencia, pero ese no fue el caso.
Más tarde, Mooreet al. se demostró que en células cultivadas con la misma mutación que causa la proteína p53 truncada, esta proteína truncada entró en el núcleo y se colocalizó con la p53 normal. También estudiaron la vida media de la proteína p53 dentro de las células y encontraron que los heterocigotos con una copia del mutante p53 tenían un aumento de tres veces en la estabilidad de la proteína p53 normal, en comparación con la estabilidad en el tipo salvaje solo, lo que significa que la estabilidad de la proteína se mejoró sobre lo normal. Estos resultados en células cultivadas extendidas y confirmaron el estudio realizado por Tyner et al. en ratones. Además, esta fue la primera vía de respuesta de división celular que se descubrió que funcionaba a través de p53 y modulaba claramente tanto la incidencia del cáncer como el envejecimiento.
En la vía Rb, que puede indicar la salida del ciclo de división celular, eventos como daño en el ADN o replicación insuficiente que conduce a telómeros cortos en los extremos de los cromosomas causan una disminución de la señalización de CDK. Esto, inturn, aumenta la actividad de la proteína quinasa Rb y, en consecuencia, aumenta la actividad del factor de transcripción E2F. ¿Cuál es el significado de E2F? Este factor de transcripción se une a los promotores de las subunidades de ARN polimerasa y a otras proteínas necesarias para que comience la fase S, y ayuda a iniciar la división celular (Campisi 2003; Weinberg 1995). Por lo tanto, parece que tanto las vías p53 como las Rb inciden en los mismos mecanismos de control del ciclo celular.