
Introducción
Las secuencias completas del genoma de varios organismos, incluidos los mamíferos, han estado disponibles recientemente como consecuencia de los rápidos avances en la tecnología de secuenciación de ADN. Sin embargo, particularmente en mamíferos, el análisis de las transcripciones todavía juega un papel clave para cerrar la brecha entre el genoma y el proteoma. Esto se debe principalmente a que, en la actualidad, no podemos predecir con precisión las estructuras de las transcripciones derivadas de un gen en particular a partir de la información genómica por sí sola. Por lo tanto, como método para el análisis de transcripciones, la construcción de bibliotecas de ADNc es crucial, incluso en la era de la secuenciación postgenómica. Aunque la clonación de ADNc de genes de interés por PCR ha proporcionado una ruta alternativa simplificada para analizar estructuras de transcripción sin la construcción de una biblioteca de ADNc, la construcción de una biblioteca de ADNc es el enfoque de elección cuando se va a analizar un gran número de ADNc de una sola fuente de ARNm. Hasta la fecha, se ha hecho un gran esfuerzo en el desarrollo de un método para la preparación de bibliotecas de ADNc de alta calidad (1-4). En contraste, la cuestión de cómo preparar una biblioteca de ADNc de alta calidad a partir de un pequeño grupo de ARN no se ha abordado tan activamente. Sin embargo, esto se ha convertido en un objetivo de alta prioridad porque los investigadores a menudo están interesados en genes hipotéticos que se predice que se expresarán solo en ciertos tipos de células o tejidos en condiciones particulares, como las que se ven en muestras patológicas.
Ha habido una serie de informes que describen el uso de pequeñas cantidades de ARN fuente para generar ADNc amplificado o ARN, a partir de los cuales se pueden preparar objetivos para el análisis de microarrays (5-15). Su objetivo final es obtener ARN de longitud completa, no sesgado en la población, que sea altamente representativo del ARNm original. Para realizar esto, estos métodos generalmente adoptan PCR o transcripción in vitro por ARN polimerasa T7 para amplificar el ARNm original en forma de ADNc o ARN, respectivamente. Aunque la comparación de perfiles de expresión es uno de los enfoques más eficientes para investigar las diferencias en los estados fisiológicos de células o tejidos, la caracterización estructural de transcripciones (por ejemplo, identificación de patrones de empalme alternativos, sitio de inicio de transcripción y sitio de terminación de transcripción) no se puede hacer mediante un análisis de microarray. El análisis secuencial de cada transcripción es inevitable en estos casos.
Desarrollamos un método que permite utilizar una pequeña cantidad de ARN inicial para construir una biblioteca de ADNc adecuada para la clonación de genes y el análisis de secuenciación integral. El método etiqueta los extremos 3′ y 5′ de los ARN de primera ronda con secuencias promotoras de fagos T7 y SP6, respectivamente, para minimizar los efectos de sesgo de tamaño durante la amplificación. Después de la amplificación de dos rondas de ARN, convertimos los ARN amplificados en ARN CDN, clonamos estos productos en un plásmido y luego evaluamos la biblioteca de ADNc resultante en comparación con la construida por un método convencional (4,16,17).
Materiales y métodos
Amplificación del ARNc
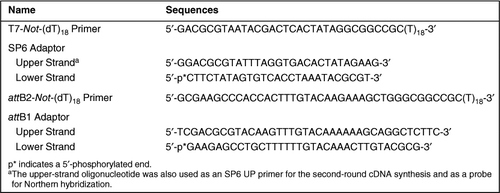
Las secuencias de oligonucleótidos utilizadas en este estudio se muestran en la Tabla 1. Para establecer el protocolo, se utilizó como plantilla 1 µg de ARN total preparado a partir de cerebro de ratón ICR (ratones machos de 8 semanas de edad). Se incubó una mezcla (10 µL) de 1 µg de ARN total y 100 pmol de imprimación T7-Not-(dT)18 a 70°C durante 10 minutos y luego se enfrió a presión sobre hielo. La síntesis de ADNc de doble cadena y la ligadura adaptadora se realizaron siguiendo los protocolos del Sistema de Plásmidos SUPERSCRIPT® (Invitrogen, Carlsbad, CA, EE. UU.) (4,16,17), con modificaciones menores. Brevemente, para la síntesis de ADNc de la primera hebra, se agregaron 4 µL 5× tampón de la primera hebra (Invitrogen), 1 µL 0,1 M ditiotreitol (TDT), 1 µL dNTPs de 10 mM, 1 µL (40 U) RNaseOUT™ (Invitrogen) y 1 µL de agua a la mezcla de imprimación desnaturalizada ARN/T7-Not-(dT)18 y se incubaron a 37°C durante 3 minutos para recocidar los cebadores. Luego, se agregaron 2 µL (400 U) de SUPERÍNDICE III RNasa H-transcriptasa inversa (Invitrogen) a la mezcla de reacción, y la temperatura se ajustó a 50°C para iniciar la síntesis de ADNc de la primera hebra. Después de 1 h, se llevó a cabo la síntesis de ADNc de segunda hebra como se describió anteriormente por Gubler y Hoffman (18); a la mezcla de ADNc de primera hebra, se agregaron 91 µL de agua, 30 µL de tampón de segunda hebra de 5× (Invitrogen), 3 µL de dNTPs de 10 mm, 1 µL (10 U) de Escherichia coli DNA ligasa, 4 µL (40 U) de E. coli DNA polimerasa y 1 µL (2 U) de E. coli RNasa H, y la mezcla se incubó a 16°C durante 2 h. Los terminales del ADNc se pulieron con 10 U T4 de ADN polimerasa (Invitrogen) a 16°C durante 5 min, y la reacción se detuvo mediante la adición de 10 µL 0,5 M de ácido etilendiamina tetraacético (EDTA). Los ARNC resultantes se purificaron por extracción con fenol / cloroformo / alcohol isoamílico (25:24:1), seguido de precipitación de etanol como se describió anteriormente (17), a excepción del uso de 1 µg de ARNt de levadura en lugar de ácido poliadenílico como portador. El ADNc precipitado se disolvió en 50 µL de TE (10 mM de Tris-HCl, pH 8,0 y EDTA de 1 mM), se mezcló con 30 µL de una solución de NaCl de polietilenglicol (PEG) 6000/2, 5 M al 20% (p/v) y se incubó en hielo durante 1 h (17). Después de la incubación, la mezcla de ADNc se centrifugó a 18.000× g a 4°C durante 15 min. El gránulo de ADNc resultante se enjuagó dos veces con etanol al 70%, se secó y luego se resuspendió en 30 µL de agua. Los ARNC purificados se ligaron con un adaptador SP6 de 500 pmol (Tabla 1) utilizando ligasa de ADN T4 de 5 U (Invitrogen) en un volumen de reacción de 50 µL a 16°C durante la noche. La mezcla unida por el adaptador se diluyó dos veces con agua, y luego los ARND se purificaron utilizando una columna DNAclear™ (Ambion, Austin, TX, EE. UU.). El eluato (en agua de 16 µL) se utilizó como plantilla para la síntesis de ARNc. Usando el kit MEGAscript ® T7 (Ambion), los cRNAs se sintetizaron a 37°C durante la noche en un volumen de reacción de 40 µL. Después de que los ARNC de plantilla se degradaron por 4 U DNasa I, los ARNc sintetizados se purificaron utilizando un Mini Kit RNeasy ® (QIAGEN, Valencia, CA, EE. UU.) y se elutaron con 100 µL de agua. La concentración del cRNA se determinó por absorción ultravioleta (UV). Para la amplificación de cRNA de segunda ronda, se utilizaron 2 µg de cRNA sintetizado y 100 pmol de cebador SP6 UP (igual al oligonucleótido de la hebra superior del adaptador SP6 que se muestra en la Tabla 1) como plantilla y cebador, respectivamente. La síntesis de ADNc de segunda ronda se llevó a cabo como en la transcripción inversa original del ARN fuente, excepto que el recocido de cebador SP6 UP se realizó a 50°C durante 3 min. Después de la purificación del ADNc de doble cadena obtenido a través de una columna de DNAclear, se utilizaron 0,5 µg de ADNc como plantilla para la siguiente amplificación de ARN asistido por polimerasa SP6. Utilizando el kit MEGAscript SP6 (Ambion), la síntesis de ARNc se llevó a cabo a 37°C durante 6 h. Después de degradar el ADNc de la plantilla utilizando DNasa I, los ARNc sintetizados se purificaron como se describió anteriormente.
Construcción de la Biblioteca de ADNc
Dos microgramos de los ARN resultantes se sometieron a síntesis de ADNc de doble cadena utilizando cebador attB2-Not-(DT)18 de 100 pmol (Cuadro 1). Los pasos de síntesis de ADNc, embotamiento final, purificación y ligadura de adaptador se llevaron a cabo como en las síntesis de ADNc anteriores, excepto que el adaptador attB1 (Tabla 1, 500 pmol) se ligó a los ADNc de doble cadena. Después de purificar los ARNC ATB1 unidos por adaptador mediante tratamientos sucesivos de extracción de fenol/cloroformo/alcohol isoamílico, precipitación de etanol y precipitación PEG/NaCl en este orden, se disolvieron en 50 µL de ET y se trataron con RNasa A (a una concentración final de 10 µg/ml; Invitrogen) para degradar el ARN contaminado a 37°C durante 30 min. La mezcla de reacción se purificó de nuevo por extracción con fenol / cloroformo / alcohol isoamílico, seguido de precipitación de etanol y precipitación de PEG/NaCl. Los ARNC resultantes se disolvieron en 15 µL de TE, y su cantidad se estimó a partir de la intensidad de tinción fluorescente después de la electroforesis en gel de agarosa (4). Los ARNC ligados a attB1 (36 ng) se sometieron a una reacción de recombinación in vitro (Sistema Gateway®; Invitrogen) con 250 ng de vector donante attP-pSP73 como se describió anteriormente (4,16,17). En resumen, el ADNc ligado a attB1 y el vector donante attP-pSP73 se mezclaron e incubaron en un volumen de reacción de 10 µL que contenía 2 µL de tampón de reacción de 5× BP Clonasa™ y 2 µL de Clonasa BP (ambos de Invitrogen) a 25°C durante la noche. La mezcla de ADNc se trató con proteinasa K de 2 µg (Invitrogen) a 37°C durante 10 minutos para apagar la reacción y se extrajo con fenol/cloroformo/alcohol isoamílico, seguido de precipitación de etanol con ARNt de levadura de 2 µg como portador. El ADNc precipitado se disolvió en 10 µL de agua, y se utilizó 1 µL de la mezcla para la transformación de células de E. coli ElectroMax™ DH10B™ (Invitrogen). Después de la titulación de la biblioteca de ADNc resultante, los plásmidos de ADNc se recuperaron de aproximadamente 1,000,000 de colonias mediante un método de dodecil sulfato de sodio alcalino (SDS) como se describió anteriormente (4,19). Los clones de ADNc resultantes estaban en una forma de plásmido que transportaba sitios ATL. Debido a que los plásmidos que transportan sitios attB son necesarios en algunas aplicaciones, estos plásmidos se convirtieron en aquellos que transportan sitios attB como se describió anteriormente (16). En consecuencia, esta biblioteca de ADNc fue designada como MB-AL (biblioteca derivada de ARN amplificado de cerebro de ratón).
De la misma manera descrita para MB-AL, construimos una biblioteca de ADNc a partir de ARN no amplificado (ARN total del cerebro de ratón de 100 µg) y la designamos como MB-CL (biblioteca de construcción convencional del cerebro de ratón).
Examen del Efecto de Sesgo de Tamaño en los ARN Amplificados
Para examinar el efecto de sesgo de tamaño en los ARN amplificados, los ARN de primera ronda transcritos por ARN polimerasa T7 y los ARN de segunda ronda transcritos por ARN polimerasa SP6 se ejecutaron en gel de agarosa al 1,0% (1 µg cada uno) y luego se secaron en una membrana de nylon (Membrana de nylon Biodyne® B; PALL, East Hills, NY, EE.UU.). Se hibridaron con cebador SP6 UP marcado con 32P (Tabla 1) y oligonucleótido No-(dT)18 (5′-GCGGCCGCTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTT-3′), respectivamente. Diez picomoles de cada sonda fueron marcados con ATP (aproximadamente 3000 Ci / mmol; Amersham Biosciences, Piscataway, NJ, EE.UU.) mediante el uso de la polinucleótido quinasa T4 (Takara, Kyoto, Japón). Después de la hibridación nocturna en tampón GMC (20) a 45°C, estas membranas se lavaron extensamente en solución de citrato salino estándar (SSC)/SDS al 1% tres veces a temperatura ambiente. El análisis de señales hibridadas se realizó en un Analizador de imágenes BAS 2000 (Película Fotográfica Fuji, Tokio, Japón)
Evaluación de Bibliotecas de ADNc
electroforesis en gel. La distribución de los tamaños de insertos de ADNc se examinó mediante electroforesis de ARN sintetizados por ARN polimerasa T7 utilizando como plantillas los ADNC plásmidos MB-AL y MB-CL no digeridos. Después de la purificación con un Mini Kit RNeasy, los ARN se electroforizaron en un gel de agarosa que contenía formaldehído con Marcadores de ARN Perfectos (EMD Biosciences, Madison, WI, EE.UU.). Después de teñir el gel con SYBR® Green II (Invitrogen), se analizó la intensidad de tinción fluorescente utilizando el software ImageQuant® (versión 5.0) en un FluorImager 595 (Amersham Biosciences).
Análisis de secuenciación aleatoria de una sola pasada. Las secuencias de 3 ‘ de clones de ADNc se examinaron mediante secuenciación de ADN. El ADN plásmido de 768 clones seleccionados al azar se purificó con un MFX-9600 Magnia® (Toyobo, Osaka, Japón) y se analizó con un secuenciador capilar RISA 384 (Shimadzu, Kyoto, Japón) (16). Después de recortar la secuencia vectorial con el Software Sequencher™ versión 4.1 (Software Hitachi, Tokio, Japón), las secuencias de ADNc obtenidas que tenían más de 200 residuos de nucleótidos se agruparon con entradas de ADNc en la base de datos GenBank® y en nuestra base de datos interna de ratones. El examen de la integridad de los extremos 3′ de los clones de ADNc se realizó mediante un análisis de si se encontró o no un hexámero de señal de poliadenilación canónica (5′-AATAAA-3’) o sus variantes de base única (11 hexámeros de señal) dentro de los 50 residuos de nucleótidos aguas arriba de la cola de poli(A) de los ADNc (21).
Microarray de ADNc. Se realizaron análisis de microarrays para comparar poblaciones de ADNc en las bibliotecas MB-AL y MB-CL. Se prepararon microarrays caseros de nailon con 3534 sondas y se realizó detección radioisotópica (22). Brevemente, los plásmidos de ADNc, que se aislaron en nuestro instituto y para los que ya se conocían secuencias completas o secuencias finales, se detectaron utilizando GeneTAC™ RA1 (Soluciones Genómicas, Ann Arbor, MI, EE.UU.) en una membrana de nylon Biodyne B y luego se fijaron siguiendo las instrucciones del fabricante. La lista de ADNC inmovilizados en este microarray está disponible a petición de los autores. Los ARNC objetivo MB-AL y MB-CL se prepararon utilizando transcripción inversa de cada una de las muestras de ARNc de 1 µg utilizadas para el análisis del tamaño del inserto como se describió anteriormente. Estos objetivos se sintetizaron y etiquetaron en la mezcla de reacción que contenía 1 µg de cada cRNA, 100 ng de hexámero aleatorio, 1× tampón de primera hebra, TDT de 10 mM, dATP, dTTP y dGTP de 800 µM, dCTP de 800 nM, dCTP de 5 µL (>2500 Ci/mmol; Amersham Biosciences) y 100 U de SuperÍndice II RNasa H inversa transcriptasa a temperatura ambiente durante 10 minutos y luego a 42°C durante 1 h. Después de la lisis alcalina y la neutralización, los ARNC etiquetados se purificaron con el kit de purificación de PCR QIAquick® (QIAGEN) y se contaron. El ADNc objetivo se sometió a hibridación con el microarray de nailon ADNc en presencia de 1 µg de ácido poliadenílico y 2,5 µg de Mouse Cot-1 DNA® (Invitrogen) en 250 µL de tampón PerfectHyb™ (Toyobo) a 68°C durante la noche. Después de un lavado riguroso a 68°C con 2× SSC/1% SDS (dos lavados de 15 min cada uno) y luego con 0,1× SSC/1% SDS (dos lavados de 30 min cada uno), se detectaron y analizaron las señales de hibridación en un FLA-8000 (Película fotográfica Fuji). Los puntos de ADNc que mostraban señales más fuertes que las del control de fondo se seleccionaron y se sometieron a análisis adicionales. Después de llevar a cabo la normalización global, se obtuvieron gráficos de dispersión de intensidades de señal de puntos de ADNc originarios de objetivos MB-AL y MB-CL.
RNA blot, hibridación. Se llevó a cabo la hibridación por blot de ARN para examinar los efectos de sesgo de tamaño. Los ARN MB-AL y MB-CL (cada uno de 1,5 µg, sintetizados como se describió anteriormente) se electroforizaron en gel de agarosa que contenía formaldehído y se transfirieron a la membrana de nylon Biodyne B. Los ARN de sonda se prepararon utilizando amplificación de PCR. Los cebadores se diseñaron en base a las secuencias registradas en la base de datos de GenBank o en nuestra base de datos interna; las plantillas eran los plásmidos de ADNc que habíamos aislado. La sonda de proteína S6 ribosómica tenía una longitud de 724 pb, amplificada con cebadores 5′-CGCTCGGCTGTGTCAAGATG-3′ y 5′-GAGGACAGCCTACGTCTCTGG-3′, y la sonda de proteína de choque térmico (hsp) de 70, 940 pb de longitud, se amplificó con 5′-GATGGACAAGGCGCAGATCC-3′ y 5′-Cebadores CTCGATGGGGGGTCCTGAGC-3′. Estas sondas se etiquetaron con dCTP (aproximadamente 3000 Ci/mmol; Amersham Biosciences) utilizando el Sistema de Etiquetado de ADN RadPrime (Invitrogen). Después de la hibridación nocturna en tampón PerfectHyb a 65°C, las membranas se lavaron sucesivamente con 0,1× SSC/1% SDS a temperatura ambiente durante 5 minutos y 15 minutos, y luego a 65°C durante 30 minutos. Las señales de hibridación se detectaron en un analizador de imágenes BAS 2000.
Resultados y discusión
El método de construcción de la biblioteca de ADNc que desarrollamos en este estudio se muestra en la Figura 1 (los pasos recién introducidos se resaltan con una estrella). Este método consiste en dos rondas de paso de amplificación de cRNA, conversión de cRNAs a cDNAs y clonación recombinacional en un plásmido. El paso clave en este método es la ligadura del adaptador para marcar cDNA termina con la secuencia del promotor SP6. Una vez que podemos sintetizar con éxito los CDNAS en este formato, se hace posible transcribir específicamente los CRNAS de primera ronda que contienen la secuencia del promotor SP6 en su extremo de 3′. Los ARNC de segunda ronda se sintetizan con ARN polimerasa SP6 utilizando los ARNC de doble cadena resultantes como plantillas. Después de las dos rondas de amplificación asistida por ARN polimerasa, los ARN resultantes se convierten en ARN de doble cadena mediante transcripción inversa utilizando el cebador attB2-Not-(dT)18. El uso de cebador attB2-Not-(dT)18 en la transcripción inversa nos permite convertir solo los cRNAs que llevan la secuencia 5′-(A)18GCGGCCGC-3′ en sus extremos 3′. Debido a que estas modificaciones solo deben permitir la amplificación y la clonación de cDNAs que tienen extremos correctamente marcados, esperamos que este método minimice el sesgo de tamaño durante la amplificación asistida por ARN polimerasa.

Se ilustran los pasos de la amplificación del ARNc asistida por ligadura de adaptador y la siguiente construcción de la biblioteca de ARNc. Para mantener los tamaños amplificados de ADNc con los de los ADNc de primera ronda, introdujimos algunas modificaciones en los métodos convencionales asistidos por ARN polimerasa (resaltados con estrellas). Las líneas azules y rojas indican ADNc y ARN, respectivamente. E. coli, Escherichia coli.
Para probar la eficacia del método, construimos bibliotecas de ADNc por un método convencional (16) utilizando ARN total no amplificado del cerebro de ratón (100 µg) y por el método descrito anteriormente utilizando 1 µg de ARN total del cerebro de ratón. La Tabla 2 resume las cantidades de ADNc y ARN obtenidas en cada paso, calculadas como un promedio de tres corridas experimentales independientes de construcción de MB-AL. Se obtuvo un promedio de 6,4×107 clones independientes de ADNc como salida final. Debido a que este número de clones de ADNc se deriva de un total de 36 ng de ADNc generados utilizando el protocolo de amplificación, calculamos que este método produce aproximadamente 1,2×1011 clones de ADNc a partir de 1 µg de ARN total.

Para examinar experimentalmente el efecto de sesgo de tamaño durante los pasos de amplificación, a continuación comparamos las distribuciones de tamaño del ARNc de primera y segunda ronda mediante análisis de hibridación por transferencia de ARN. En estos experimentos, los ARN de primera y segunda ronda se hibridaron con cebador SP6 UP y con oligonucleótido No-(dT)18, respectivamente, porque estas sondas de oligonucleótidos solo detectan ARN transcritos correctamente hasta el final. Como se muestra en la Figura 2, se encontró que el tamaño del ARN en el pico de la señal de hibridación en el ARN de segunda ronda era casi el mismo que en el ARN de primera ronda, pero con un ligero cambio a un tamaño más pequeño. Sospechamos que el truncamiento de los ARN probablemente tuvo lugar durante la segunda ronda de síntesis de ADNc con el cebador SP6 UP.

para examinar el efecto de sesgo de tamaño en los ARN. Se trazaron señales de hibridación a lo largo de la dirección de la electroforesis de cRNA. El tamaño del ARN se determinó con base en la movilidad del marcador de escalera de ARN. Línea azul, la primera ronda de cRNA hibridada con sonda SP6 UP; línea roja, la segunda ronda de cRNA hibridada con sonda Not-(dT)18; valor PSL, intensidad de señal indicada arbitrariamente por el Software de Medición de Imágenes (Película fotográfica Fuji).
Para evaluar aún más la calidad de la biblioteca de ADNc amplificada resultante, también comparamos varias características de las bibliotecas de ADNc generadas con y sin la amplificación (MB-AL y MB-CL, respectivamente). En primer lugar, examinamos la distribución de tamaños de los insertos de ADNc, como se muestra en la Figura 3A. Los resultados de las señales de intensidad de fluorescencia obtenidas de la imagen de tinción de gel indicaron que los tamaños de insertos de ADNc de MB-AL y MB-CL eran similares, pero el punto máximo era ligeramente menor en MB-AL (0,65 kb) en comparación con el de MB-CL (0,8 kb). A continuación, analizamos secuencias de 3 ‘ de clones de ADNc muestreados aleatoriamente de cada biblioteca. Los resultados de agrupamiento de estas etiquetas de secuencia expresada (EST) se muestran en la Figura 3B y sugieren que la complejidad de MB-AL era tan alta como la de MB-CL. Sin embargo, vale la pena señalar que los clones de ADNc altamente redundantes desaparecieron en MB-AL. Mediante un análisis estadístico (prueba Chi), se demostró que la diferencia de redundancias entre MB-AL y MB-CL era significativa (la probabilidad de que las redundancias en MB-AL y MB-CL fueran similares es <0,001). Además, examinamos la integridad de los extremos 3’ de los cDNAs analizando si cada EST contenía un hexámero de señal de poliadenilación o no. Los resultados indicaron que el 80,1% y el 87,3% de los clones de ADNc en MB-AL y MB-CL, respectivamente, contenían secuencias plausibles de señales de poliadenilación (21). La disminución de la tasa de ocurrencia de hexámeros de señal de poliadenilación en MB-AL probablemente indicó un aumento en el número de clones de ADNc donde la síntesis de ADNc se imprimó internamente debido a dos rondas de cebado dT.

MB-AL (biblioteca derivada de cRNA amplificada de cerebro de ratón) se evaluó en comparación con una biblioteca construida convencionalmente, MB-CL. A) Comparación de las longitudes de ADNc de inserción. Los ARN transcritos in vitro se fraccionaron por tamaño en gel de agarosa y se teñieron. Sus intensidades fluorescentes a lo largo de la dirección de la electroforesis (indicadas como tamaño de ARN) se muestran en unidades fluorescentes relativas (rfu). B) Resultados de agrupamiento de secuencias de final de 3’MB-AL y MB-CL (EST). C) Comparación de la población de ADNc en MB-AL y MB-CL. Se trazaron las señales obtenidas por hibridación de microarrays. (D) Los resultados de la hibridación por blot de ARN sobre la proteína S6 ribosómica y la hsp 70. Los ARN MB-AL estaban en el carril izquierdo y los de MB-CL en el carril derecho (indicados por AL y CL, respectivamente). Las puntas de flecha indican las posiciones del tamaño de ARN previsto de cada gen. hsp, proteína de choque térmico.
Mediante hibridación por microarray, examinamos la población de ADNc de MB-AL en comparación con la de MB-CL. La Figura 3C muestra un gráfico de dispersión de señales de hibridación de objetivos MB-AL y MB-CL. Los resultados indicaron una correlación considerablemente alta de señales de hibridación entre objetivos MB-AL y MB-CL, lo que sugiere que no hubo un efecto perjudicial de la amplificación en la población de ADNc.
Finalmente, se realizó un análisis de hibridación por transferencia de ARN para examinar los efectos de sesgo de tamaño con atención a los genes de limpieza (Figura 3D). Para la proteína S6 ribosómica y la hsp 70, los tamaños de ARNc en MB-AL y MB-CL fueron similares y comparables a los esperados de las secuencias de nucleótidos notificadas. Sin embargo, el patrón de hibridación de los ARN hsp 70 en MB-AL fue ligeramente diferente del de MB-CL, lo que indica que el efecto de sesgo de tamaño no se eliminó por completo.
El método de construcción de la biblioteca de ADNc descrito en este informe fue diseñado para minimizar los sesgos de tamaño durante los pasos de amplificación asistidos por ARN polimerasa. Tal método de construcción de biblioteca asistida por ARN polimerasa a partir de un pequeño grupo de ARN para un análisis de secuenciación integral no ha sido bien estudiado, mientras que se han buscado activamente métodos de construcción de biblioteca de ADNc/ARN para hibridación de microarrays. Aunque Lukyanov et al. (23) y Piao et al. (24) métodos reportados que utilizan amplificación de PCR para construir una biblioteca de ADNc a partir de una cantidad submicrograma de ARN total, la amplificación de PCR tiene intrínsecamente desventajas como un sesgo de población y tamaño severo y baja fidelidad de la amplificación de ADNc. Por eso, en este estudio, intentamos desarrollar un método de construcción de una biblioteca de ADNc basado en la amplificación asistida por ARN polimerasa.
Para este propósito, diseñamos una nueva estrategia de amplificación asistida por ARN polimerasa, como se muestra en la Figura 1, debido a los métodos de Eberwine et al. (5), Huang et al. (10), y Lin et al. (11) todavía tienen desventajas para la preparación de un ADNc contenido en plásmidos adecuado para un análisis exhaustivo de la estructura génica. En el primero, la síntesis de ADNc después de la amplificación del ARN se realiza con hexámeros aleatorios, y esto hace que los ADNc amplificados sean inevitablemente más cortos que los originales (5,7). En este último caso, la introducción de una cola homopolimérica en el extremo de la primera cadena del ADNc por la actividad de la transcriptasa inversa de la desoxinucleotidil transferasa (TdT) terminal o del virus de la leucemia murina Moloney (MMLV) tiene sentido para minimizar el efecto de sesgo de tamaño durante la amplificación asistida por ARN polimerasa, como en nuestro método. Sin embargo, se sabe que el método de cola homopolimérica causa el truncamiento de los ADNc debido al cebado inesperado de la síntesis de ADNc a partir de un estiramiento homopolimérico de secuencias internas de ARN. Debido a que el método de ligadura por adaptador descrito en este estudio podría proporcionar una secuencia específica para el cebado de ADNc, podríamos esperar reducir el grado de truncamiento de ADNc utilizando nuestro método.
En principio, nuestro método solo debe amplificar los ADNc flanqueados por una cola poli(A) intrínseca y la secuencia adaptadora SP6, que se generaron en la primera ronda de síntesis de ADNc. Los resultados mostrados en la Figura 2 y la Figura 3A fueron esencialmente consistentes con esto, aunque la población de ADNc en el rango de tamaño más pequeño aumentó un poco. Los resultados del análisis de ARN blot (Figura 3D) también mostraron que el efecto de sesgo de tamaño podría reducirse, pero no eliminarse por completo, incluso en nuestro método. Esto se debió probablemente a que los ARNC truncados adjuntos a una cola poli (A) y la secuencia promotora SP6 se generaron durante la conversión de ARNc a ADNc. Esta opinión fue apoyada por el hecho de que la tasa de ocurrencia de las secuencias de señales de poliadenilación en MB-AL fue significativamente menor que la observada en la biblioteca convencional de ADNc (21). El truncamiento de los ADNc podría ocurrir durante la síntesis de ADNc debido al cebado interno del ARN con la imprimación de cola dT y la imprimación SP6. Debido a que los estiramientos homopolímeros ocurren con frecuencia en ARNm eucarióticos, el método de relaves homopolímeros podría imponer un sesgo de tamaño más severo en la amplificación de ARNc de múltiples rondas que nuestro método de ligadura por adaptador. Aunque el aumento de la temperatura de reacción durante la transcripción inversa y la disminución de la concentración de imprimación podrían suprimir el cebado interno aberrante en la síntesis de ADNc hasta cierto punto, se sabe que es difícil eliminarlo por completo en la actualidad. Por lo tanto, aunque realizamos una amplificación de dos rondas del ARN para la demostración del protocolo general, en la práctica, recomendamos omitir la amplificación de la segunda ronda del ARN cuando se puede obtener suficiente ARN en la reacción de la primera ronda.
Considerando estos resultados, concluimos que nuestro nuevo método es útil para la construcción de bibliotecas de ADNc a partir de una pequeña cantidad de ARN inicial. De hecho, hemos utilizado con éxito y de forma rutinaria este método para la construcción de bibliotecas de ADNc cuando la cantidad de ARN total es inferior a 1 µg (datos no mostrados). Debido a que 1 µg de ARN total se puede recuperar de 105-106 células de mamíferos o 1 mg de tejidos de mamíferos, las bibliotecas de ADNc se pueden construir fácilmente a partir de células fraccionadas mediante clasificadores celulares o microdisección utilizando nuestro método. Además, debido a que calculamos que este método nos permite obtener más de 105 clones de ADNc a partir de 1 pg de ARN total, que es cercano a la cantidad de ARN total en una sola célula, se podría construir una biblioteca de ADNc derivada de una sola célula basada en esta amplificación de ADNc asistida por ligadura de adaptador.
Agradecimientos
Los autores agradecen la instrucción de análisis estadístico del Dr. Kazuharu Misawa; la división de ADN de plásmidos del Dr. Hisashi Koga; y la ayuda técnica de la Sra. Akiko Ukigai-Ando y el Sr. Takashi Watanabe. El estudio fue apoyado por una subvención del Instituto de Investigación de ADN de Kazusa a R. O., R. F. K. y O. o.
Declaración de intereses en conflicto
Los autores declaran no tener intereses en conflicto.
- 1. Carninci, P., C. Kvam, A. Kitamura, T. Ohsumi, Y. Okazaki, M. Itoh, M. Kamiya, K. Shibata, et al.. 1996. Clonación de ADNc de larga duración de alta eficiencia por trampero con TAPA biotinilada. Genomics 37: 327-336.Crossref, Medline, CAS, Google Scholar
- 2. Ohara, O., T. Nagase, K. Ishikawa, D. Nakajima, M. Ohira, N. Seki, and N. Nomura. 1997. Construcción y caracterización de bibliotecas de ADNc del cerebro humano adecuadas para el análisis de clones de ADNc que codifican proteínas relativamente grandes. DNA Res. 4: 53-59.Crossref, Medline, CAS, Google Scholar
- 3. Suzuki, Y., K. Yoshitomo-Nakagawa, K. Maruyama, A. Suyama, and S. Sugano. 1997. Construcción y caracterización de una biblioteca de ADNc enriquecida de longitud completa y una biblioteca de ADNc enriquecida de 5 pies. Gene 200:1149-1156.Crossref, Google Scholar
- 4. Ohara, O. and G. Temple. 2001. Construcción de la biblioteca direccional de ADNc asistida por la reacción de recombinación in vitro. Ácidos nucleicos Res.29:e22.Crossref, Medline, CAS, Google Scholar
- 5. Eberwine, J., H. Yeh, K. Miyashiro, Y. Cao, S. Nair, R. Finnel, M. Zettel, and P. Coleman. 1992. Análisis de la expresión génica en neuronas vivas individuales. Proc. Natl. Acad. Sci. USA 89: 3010-3014.Crossref, Medline, CAS, Google Scholar
- 6. Wang, E., L. D. Miller, G. A. Ohnmacht, E. T. Liu, and F. M. Marincola. 2000. Amplificación de ARNm de alta fidelidad para perfiles genéticos. NAT. Biotechnol. 18:457–459.Crossref, Medline, CAS, Google Scholar
- 7. Baugh, L. R., A. A. Hill, E. L. Brown, and C. P. Hunter. 2001. Análisis cuantitativo de amplificación de ARNm por transcripción in vitro. Ácidos nucleicos Res.29:e29.Crossref, Medline, CAS, Google Scholar
- 8. Zhumabayeva, B., L. Diatchenko, A. Chenchik, and P. D. Siebert. 2001. Uso de ADNc generado por SMART™para estudios de expresión génica en múltiples tumores humanos. BioTechniques 30: 158-163.Link, CAS, Google Scholar
- 9. Gomes, L., R. Silva, B. S. Stolf, E. B. Cristo, R. Hirata, Jr., F. A. Soares, L. Reis, E. J. Neves, et al.. 2003. Análisis comparativo de ARN amplificado y no amplificado para hibridación en microarray de ADNc. Anal. Bioquímica. 321:244–251.Crossref, Medline, CAS, Google Scholar
- 10. Huang, J., T. Lin, D. Chang, S. Lin, and S. Ying. 2003. Bcl-2 truncado, un posible marcador pre-metástico en el cáncer de próstata. Bioquímica. Biophys. Res. Commun. 306:912–917.Crossref, Medline, CAS, Google Scholar
- 11. Lin, S.L. y S. Y. Ying. 2003. Construcción de la biblioteca de ARNm/ADNc utilizando la reacción de ciclo de ARN-polimerasa. Métodos Mol. Biol. 221:129–143.Medline, CAS, Google Scholar
- 12. Matz, M. V. 2003. Amplificación de depósitos representativos de ADNc a partir de cantidades microscópicas de tejido animal. Métodos Mol. Biol. 221:103–116.Medline, CAS, Google Scholar
- 13. Petalidis, L., S. Bhattacharyya, G. A. Morris, V. P. Collins, T. C. Freeman, and P. A. Lyons. 2003. Amplificación global de ARNm por PCR de conmutación de plantillas: linealidad y aplicación al análisis de microarrays. Ácidos nucleicos Res. 22: e142.Crossref, Google Scholar
- 14. Polacek, D. C., A. G. Passerini, C. Shi, N. M. Francesco, E. Manduchi, G. R. Grant, S. Powell, H. Bischof, et al.. 2003. Fidelidad y sensibilidad mejorada de los perfiles de transcripción diferencial tras la amplificación lineal de cantidades de nanogramas de ARNm endotelial. Fisiol. Genomics 13: 147-156.Crossref, Medline, CAS, Google Scholar
- 15. Wilson, C., S. D. Pepper, Y. Hey, y C. J. Miller. 2004. Los protocolos de amplificación introducen errores sistemáticos pero reproducibles en los estudios de expresión génica. BioTechniques 36: 498-506.Link, CAS, Google Scholar
- 16. Ohara, O., T. Nagase, G. Mitsui, H. Kohga, R. Kikuno, S. Hiraoka, Y. Takahashi, S. Kitajima, et al.. 2002. Caracterización de la biblioteca de ADNc fraccionados por tamaño generada por el método asistido por recombinación in vitro. DNA Res. 9: 47-57.Crossref, Medline, CAS, Google Scholar
- 17. Ohara, O. 2003. Construcción de una biblioteca de ADNc fraccionada por tamaño asistida por una reacción de recombinación in vitro. Métodos Mol. Biol. 221:59–71.Medline, CAS, Google Scholar
- 18. Gubler, U. y B. J. Hoffman. 1983. Un método simple y muy eficiente para generar bibliotecas de cDNA. Gene 25: 263-269.Crossref, Medline, CAS, Google Scholar
- 19. Sambrook, J. and D. W. Russell. 2001. Preparación de ADN plásmido por lisis alcalina con SDS, p. 1.31–1.42. Clonación Molecular, (3ª edición). CSH Laboratory Press, Cold Spring Harbor, NY.Google Scholar
- 20. Church, G. M. y W. Gilbert. 1984. Secuenciación genómica. Proc. Natl. Acad. Sci. USA 81: 1991-1995.Crossref, Medline, CAS, Google Scholar
- 21. Beaudoing, E., S. Freier, J. R. Wyatt, J. M. Claverie, and D. Gautheret. 2000. Patrones de uso de señales de poliadenilación de variantes en genes humanos. Genome Res. 10: 1001-1010.Crossref, Medline, CAS, Google Scholar
- 22. Tanaka, T. S., S.A. Jaradat, M. K. Lim, G. J. Kargul, X. Wang, M. J. Grahovac, S. Pantano, Y. Sano, et al.. 2000. Perfil de expresión de todo el genoma de la placenta y el embrión de gestación media utilizando un microarray de cADN de desarrollo de 15.000 ratones. Proc. Natl. Acad. Sci. USA 97: 9127-9132.Crossref, Medline, Google Scholar
- 23. Lukyanov, K., L. Diatchenko, A. Chenchik, A. Nanisetti, P. Siebert, N. Usman, M. Matz, and S. Lukyanov. 1997. Construcción de bibliotecas de ADNc a partir de pequeñas cantidades de ARN total utilizando el efecto PCR de supresión. Bioquímica. Biophys. Res. Commun. 230:285–288.Crossref, Medline, CAS, Google Scholar
- 24. Piao, Y., N. T. Ko, M. K. Lim, and M. Ko. 2001. Construcción de bibliotecas de ADNc enriquecidas con transcripciones largas a partir de cantidades submicrogramas de ARN totales mediante un método de amplificación de PCR universal. Genome Res. 11: 1553-1558.Crossref, Medline, CAS, Google Scholar