
hvorfor udvikle Cap analyse af genekspression?
forskning af Piero Carninci og Yoshihide Hayashisaki i slutningen af 1990 ‘ erne, som startede med cap trapper-metoden, anvendelse af trehalose, normaliserings – /subtraktionsmetoden og en ny kloningsvektor, satte scenen for udvikling af Cap-analyse af genekspression. Med cap trapper isoleres cDNA/mRNA-hybrider i fuld længde, og mRNA ‘ et biotinyleres kemisk på hættestrukturen, og streptavidin-belagte magnetiske perler fanger hybriderne. Dette fremskridt i serien af større DNA-teknologier beskrives af Nature som milepæl 5 (www.nature.com/milestones/miledna/full/miledna05.html). deres formål med at udvikle CAGE var at skabe en teknologi til omfattende kortlægning af langt størstedelen af humane transkriptionsstartsteder og deres promotorer. Faktisk eksisterede teknologien til at profilere aktiviteten af gentranskription på hvert promotorsted ikke, før CAGE kom på scenen. Messenger RNA (mRNA) repræsenterer en kritisk forbindelse mellem Informationen kodet i individuelle gener på et genom og protein makeup, der bestemmer en organisms skæbne. Vi spurgte os selv: Hvad er de genomiske regioner eller promotorer, der driver det specifikke udtryk for gener og deres unikt forskellige RNA ‘ er? Faktisk har cDNA-samlinger i fuld længde vist, at de fleste gener har mere end et transkriptionsstartsted, og det er derfor ret vanskeligt at identificere de kontrollerende regioner, det vil sige promotorer, der er ansvarlige for ekspressionen af de forskellige former for udskrifter. analyse af kompleksiteten af denne informationsoverførselsproces, kaldet ‘transkription’, kræver udvikling af sofistikerede molekylære værktøjer, der kan fange både de kvalitative og kvantitative aspekter af genekspression. Så med CAGE, vores originale genom-dækkende transkriptionsstartdetekteringsteknologi, kan vi udføre profilering med høj genekspression med samtidig identifikation af væv/celle/tilstandsspecifikke transkriptionsstartsteder (TSS), herunder promotoranvendelsesanalyse. CAGE er baseret på forberedelse og sekventering af sammenkædere af DNA-tags, der stammer fra de indledende 20 nukleotider fra 5′ – end mRNA ‘ er, som afspejler den oprindelige koncentration af mRNA i den analyserede prøve (RNA-frekvens).
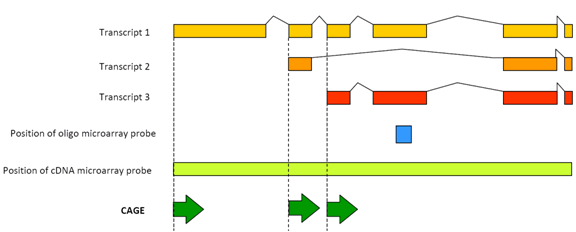
Figur 1: CAGE registrerer transkriptionsaktiviteten for hvert promotortranskript.
udtrykte sekventeringskoder (Est ‘ er) blev brugt i tidlige stadier af teknologiudvikling til at identificere promotorelementer ved at tilpasse dem til det menneskelige genom. Denne proces er imidlertid meget dyr på grund af omkostningerne ved håndtering af fysiske cDNA ‘ er og Sanger-sekventering. En måde at overvinde disse problemer på er at bruge taggingsteknologier, som er udviklet til at detektere udskrifter med en følsomhed, der er mindst en størrelsesorden større end EST-sekventering, derefter udtømmende identificere udskrifter, identificere deres promotorer og korrelere dem med ekspressionsprofiler ved at tælle tags som et digitalt mål for genekspression.
disse sekvenser (også kaldet “tags”) justeres derefter til genomsekvenser ved hjælp af enkle beregningsprocedurer (kaldet BLAST) og tælles, hvilket giver en måling af frekvensen af RNA-ekspression. Da disse sekvensmærker identificerer startstederne for RNA-transkriptionen, identificerer de også genomsekvenserne tæt på disse startsteder. De nærliggende regioner er kernepromotorregionerne, som er genomiske sekvenser, der får gener til at blive transkriberet under de mange forskellige forhold, der opstår i mange komplekse organismer, fra mus til mennesker.
attributter af CAGE
CAGE har store fordele sammenlignet med klassiske mikroarray-baserede ekspressionsdetekteringsteknikker. Faktisk ved at identificere promotoren, der forårsager RNA-transkription i hvert biologisk fænomen, væv, celle osv. vi kan identificere de DNA-regulerende elementer, der er specifikke for hvert biologisk fænomen ved at se på sekvenserne, der er i promotorerne af RNA-isoformerne, der udtrykkes i de analyserede prøver. Promotorer indeholder specifikke sekvenser eller transkriptionsfaktorbindingssteder (TFB ‘ er), der genkendes af deres bindende proteiner kaldet transkriptionsfaktorer (TF) og fremmer eller alternativt undertrykker transkription. Ved hjælp af beregningsmetoder analyserer vores forskere promotorer med lignende ekspressionsprofiler for deres TFB ‘er og identificerer derefter de TF’ er, der er ansvarlige for genomets transkriptionelle output. Ved at tælle antallet af BURMÆRKER for hver promotor inden for et gen kan vi nu ikke kun bestemme RNA-ekspressionsniveauet (dette er en digital detektion af frekvens), men vigtigere også fra hvilken af de forskellige alternative promotorer RNA transkriberes.
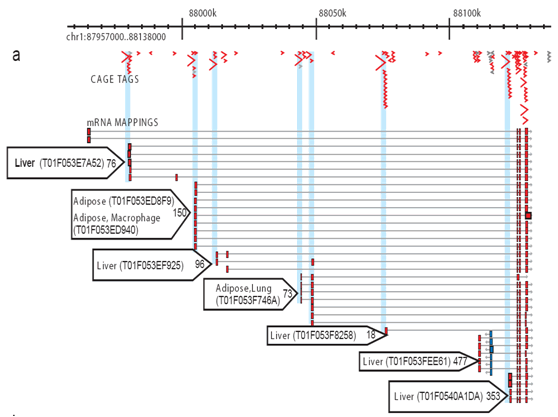
figur 2: CAGE tillader omfattende profilering af aktiveringer på hvert promotorsted. For hvert bibliotek, et antal BURMÆRKER sekventeres og justeres til genomet, så den specifikke transkriptionsaktivitet ved hver promotor kan måles, og hver promotors bidrag skelnes. Dette forenklede eksempel viser fedt-og leverkernepromotorer. Tiny blå pile: individuelle bur tags; røde pile: promotor brug præference for væv; røde kasser: core promotor regioner.
som nævnt bruger CAGE cap-fældefangst som det første skridt til at fange 5′ – enderne af cDNA ‘ erne, som derefter omdannes til kort sekvens (tags) på 20 Til 27 nt svarende til mRNA TSSs, . Vi har produceret millioner af mus-og menneskelige BURMÆRKER ved hjælp af sammenkædede BURMÆRKER med Sanger-sekventering, indtil vi for nylig flyttede til deepCAGE, som vi bruger anden generations sekventering til.
indtil 2006 kørte vi BURBIBLIOTEKER på vores oprindelige RISA-sekventeringsledning, som blev bygget i slutningen af 90 ‘ erne og omfattede den eneste kapillærsekvensator med en række 384 kapillærer, som vi udviklede i samarbejde med SHIMADU Corporation.
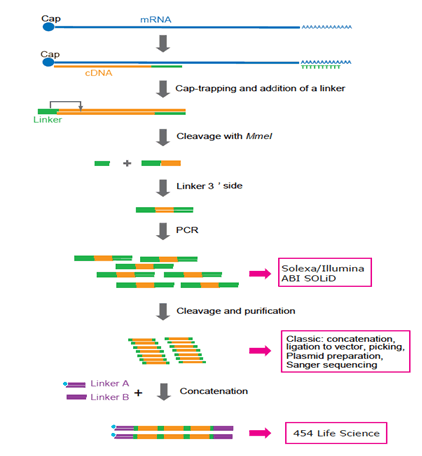
figur 3: repræsentation af BURFORBEREDELSESPROTOKOL tilpasset forskellige platforme. Soleks og Illumina foretrækkes. 454 Life Sciences bruges ikke længere, fordi sammenkædning kræver yderligere PCR-cyklusser og kompliceret manipulation. I fremtiden foretrækkes sekventeringsteknologi med et molekyle, fordi PCR muligvis ikke er påkrævet.
selvom tagging-og sekventeringsteknologi er udviklet ved hjælp af seriel analyse af genanalyse (SAGE) – metoden, er CAGE unik, fordi den er baseret på princippet om profilering af 5′-enden af RNA ‘er, der bærer en cap-site-all mRNA’ er og en stor brøkdel af de ikke-kodende RNA ‘ er. Vi har udviklet 27-nt lange BURMÆRKER for at øge kortlægningseffektiviteten.