
proč vyvinout Cap analýzu genové exprese?
Výzkum Piero Carninci a Yoshihide Hayashizaki v pozdní 1990, která začala s čepice trapper metoda, použití trehalosa, normalizaci/odčítání metody, a nový klonování vektor, připravila půdu pro rozvoj Szp Analýzu Genové Exprese. S čepice trapper, full-délka cDNA/mRNA hybridy jsou izolované, a mRNA je chemicky biotinylated na cap konstrukce a streptavidin-coated magnetic beads zachytit hybridy. Tato záloha v řadě významných DNA technologií je popsán v Přírodě jako Milník 5 (www.nature.com/milestones/miledna/full/miledna05.html). Jejich účel v rozvojových KLECE bylo vytvořit technologii pro komplexní mapování drtivá většina lidské transkripce začíná lokalit a jejich organizací. Ve skutečnosti technologie profilování aktivity genové transkripce v každém místě promotoru neexistovala, dokud CAGE nepřišel na scénu. Messenger RNA (mRNA) představuje kritické spojení mezi informacemi zakódovanými v jednotlivých genech na genomu a proteinovým make-upem, který určuje osud organismu. Ptali jsme se sami sebe: jaké jsou genomové oblasti nebo promotory, které řídí specifickou expresi genů a jejich jedinečně odlišných RNA? Kolekce cDNA v plné délce skutečně ukázaly, že většina genů má více než jedno výchozí místo transkripce, a proto je docela obtížné identifikovat kontrolní oblasti, tj.
Analýze složitosti tohoto převodu zpracovávat informace, se nazývá ‚transkripce‘, vyžaduje vývoj sofistikované molekulární nástroje, které mohou zachytit obě kvalitativní a kvantitativní aspekty genové exprese. Takže s KLECÍ, naše původní genom-wide transkripce začíná technologie detekce, můžeme udělat vysoký-v průběhu profilování genové exprese se současnou identifikací tkáně/buňky/podmínky specifické transkripční začátek stránky (TSS), včetně propagátorem využití analýzy. CAGE je založena na přípravě a sekvenování konkatamerů dna tagů pocházejících z počátečních 20 nukleotidů z 5 ‚ koncových mRNA, které odrážejí původní koncentraci mRNA v analyzovaném vzorku (frekvence RNA).
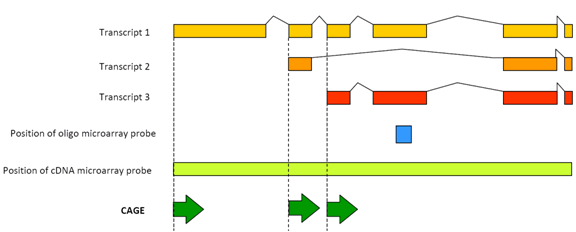
Obrázek 1: CAGE detekuje transkripční aktivitu každého přepisu promotoru.
vyjádřené sekvenační značky (est) byly použity v raných stádiích vývoje technologie k identifikaci promotorových prvků jejich zarovnáním na lidský genom. Tento proces je však velmi nákladný kvůli nákladům na manipulaci s fyzickými cDNA a sangerovým sekvenováním. Jeden způsob, jak překonat tyto problémy, je použít značkování technologií, které byly vyvinuty pro detekci transkriptů s citlivostí, která je nejméně o jeden řád větší než EST sekvenování, pak vyčerpávajícím způsobem identifikovat přepisy, identifikovat jejich organizace a jejich korelaci s výrazem profily počítáním kategorie jako digitální měření genové exprese.
Tyto sekvence (také volal „tagy“) jsou pak zarovnány k sekvencím genomů pomocí jednoduché výpočetní postupy (BLAST) a počítal, která dává měření frekvence RNA výraz. Protože tyto sekvenční značky identifikují počáteční místa transkripce RNA, identifikují také genomové sekvence blízké těmto výchozím místům. Sousední regiony jsou core promotor regiony, které jsou genomových sekvencí, které způsobují geny budou přepsány na mnoho různých podmínek, které se vyskytují v mnoha složitých organismů, z myší na lidi.
atributy klece
klec má velké výhody ve srovnání s klasickými technikami detekce exprese na bázi microarray. Ve skutečnosti identifikací promotoru, který způsobuje transkripci RNA v každém biologickém jevu, tkáni, buňce atd. můžeme identifikovat DNA regulační prvky, které jsou specifické pro každý biologický jev, při pohledu na sekvence, které jsou v předkladatelů RNA izoformy jsou vyjádřeny v analyzovaných vzorků. Pořadatelé obsahovat specifické sekvence, nebo transkripční faktor vazebná místa (TFBS), které jsou uznávány jejich vazebné proteiny nazývané transkripční faktory (TF) a podpořit, případně potlačit, transkripce. Pomocí výpočetních metod naši vědci analyzují promotory s podobnými expresními profily pro své TFB a poté identifikují TFs zodpovědné za transkripční výstup genomu. Počítáním počtu klecových značek pro každý promotor v rámci genu můžeme nyní určit nejen úroveň exprese RNA (jedná se o digitální detekci frekvence), ale také, ze kterého z různých alternativních promotorů je RNA přepsána.
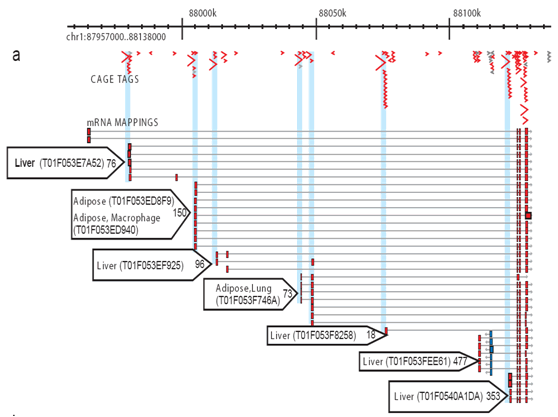
Obrázek 2: CAGE umožňuje komplexní profilování aktiv na každém místě promotoru. Pro každou knihovnu je řada značek klece sekvenována a zarovnána s genomem, takže lze měřit specifickou transkripční aktivitu u každého promotoru a rozlišit příspěvek každého promotoru. Tento zjednodušený příklad ukazuje promotory tukového a jaterního jádra. Malé modré šipky: jednotlivé značky klece; červené šipky: preference použití promotoru pro tkáně; červené boxy: hlavní oblasti promotoru.
Jak již bylo zmíněno, KLEC používá cap-zachycení jako první krok k zachycení 5′ konce cDNAs, které jsou pak transformovány do krátké sekvence (tagy), 20 až 27 nt odpovídající mRNA TSSs, . Vyrobili jsme miliony značek myší a lidských klecí pomocí zřetězených značek klecí se sekvenováním Sanger, až jsme se nedávno přestěhovali do deepCAGE, pro které používáme sekvenování druhé generace.
Do roku 2006 jsme byli běží KLEC knihoven na naše původní RISE sekvenování potrubí, která byla postavena na konci 90. let a zahrnovala pouze kapilární sekvencer s řadou 384 kapilárách, které jsme vyvinuli ve spolupráci s Shimadzu Corporation.
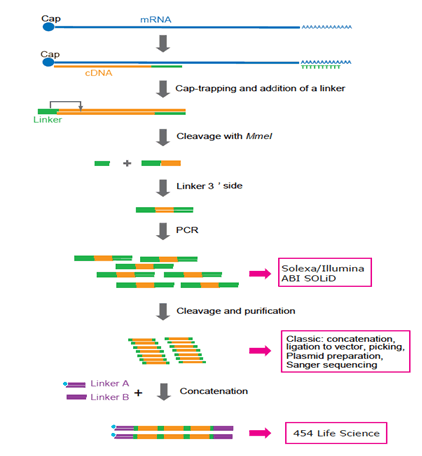
Obrázek 3: reprezentace protokolu přípravy klece přizpůsobeného různým platformám. Nyní jsou preferovány Solexa a Illumina. 454 Life Sciences (systém FLX) se již nepoužívá, protože zřetězení vyžaduje další PCR cykly a komplikovanou manipulaci. V budoucnu bude upřednostňována technologie sekvenování s jednou molekulou, protože PCR nemusí být vyžadována.
i když značkování a sekvenování technologie byla vyvinuta pomocí sériové analýzy genové analýzy (ŠALVĚJ) metoda ,, KLEC je unikátní, protože je založen na principu profilování na 5′ konci Rna nesoucí cap-site – všechny mRNAs a velký podíl non-coding RNAs. Vyvinuli jsme 27 – NT dlouhé klecové značky pro zvýšení efektivity mapování.